




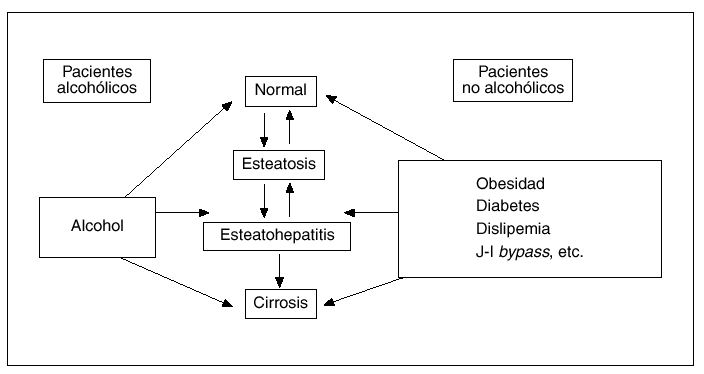



Desde un punto de vista conceptual, por esteatohepatitis (EH) se entiende la complicación necroinflamatoria de una esteatosis hepática persistente, constituyendo así la principal vía por la cual la esteatosis hepática puede progresar a cirrosis (fig. 1)1,2. Aunque en un principio se creyó que la EH era expresión exclusivamente del abuso crónico de alcohol, en 1980 Ludwig et al3 describieron una serie de pacientes que no ingerían alcohol y presentaban una enfermedad hepática caracterizada por la presencia de esteatosis, indistinguible de la que se veía en pacientes alcohólicos. Un hecho interesante fue que un número significativo de estos casos presentaba signos histológicos de inflamación caracterizados por la infiltración de neutrófilos y linfocitos tanto a nivel lobulillar como portal, lo que motivó que estos autores acuñaran el término esteatohepatitis no alcohólica (EHNA) para los casos de esteatosis hepática con signos inflamatorios en pacientes no alcohólicos. No obstante, en la actualidad un creciente número de autores consideramos que el término EH no refleja con exactitud el espectro evolutivo de este trastorno hepático que, tal como se esquematiza en la figura 1, comprende desde la esteatosis hepática simple hasta la cirrosis establecida. Por todo ello creemos que la denominación «enfermedad hepática por depósito de grasa» parece más adecuada para definir a esta entidad anatomoclínica que se reconoce cada vez con mayor frecuencia en nuestro medio. En cualquier caso, dado que el término de EHNA es el más aceptado internacionalmente, es el que utilizaré a lo largo de esta revisión, en la que se tratarán con particular detalle los factores etiológicos, los mecanismos patogénicos más recientemente implicados y las pautas recomendadas para el tratamiento de esta enfermedad hepática.
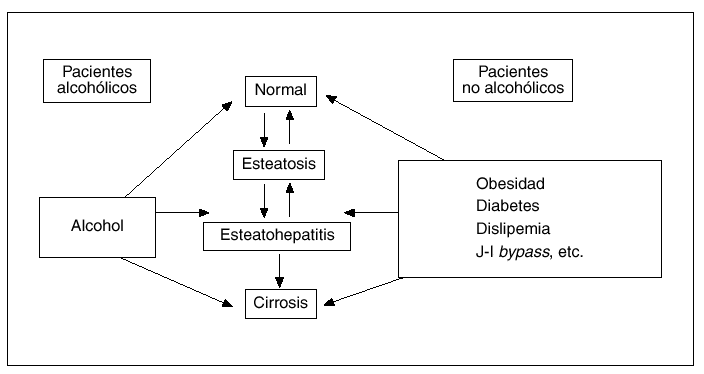
Fig. 1. Espectro evolutivo de la enfermedad hepática por depósito de grasa.
FACTORES ETIOLÓGICOS
La EHNA se asocia etiológicamente con una amplia variedad de situaciones clínicas, como la diabetes mellitus y la dislipemia (tabla I), pero es la obesidad el principal factor etiológico de esta enfermedad del hígado. En este sentido, diferentes estudios epidemiológicos retrospectivos han observado signos histológicos de EHNA en el 60-90% de la población obesa analizada4,5. Más recientemente, nuestro grupo ha publicado un estudio prospectivo sobre una población de pacientes con obesidad mórbida que ingresaron en el hospital para la realización de una gastroplastia quirúrgica6. A cada uno de estos pacientes se les realizó una biopsia hepática y pudimos observar que el 70% de estas personas, con un índice de masa corporal (IMC) medio de 50,5 kg/m2, presentaban signos histológicos de EHNA.

La diabetes mellitus, generalmente no insulinodependiente o tipo 2, es la segunda entidad, en términos de frecuencia, asociada con la EHNA1,2, y se ha encontrado en el 34-75% de los pacientes frecuentemente asociada a obesidad1,2,7. En un estudio de necropsias, Wanless et al encontraron que obesidad y diabetes estaban presentes en 20 de 22 pacientes con EHNA5, y demostraron una mayor prevalencia en pacientes con diabetes tipo 2 que requerían insulina. Por el contrario, en nuestro estudio la prevalencia de diabetes mellitus fue sólo del 19% en los pacientes obesos con EHNA analizados, aunque un hecho interesante fue que todos los pacientes diabéticos padecían EHNA6.
Un aspecto interesante que cabe destacar es que la resistencia periférica a la insulina y la consiguiente hiperinsulinemia son alteraciones metabólicas que se observan comúnmente en la diabetes mellitus tipo 2 y en la obesidad8, lo que sugiere que la resistencia tisular a la acción de la insulina podría desempeñar un importante papel en la génesis de la «enfermedad hepática por depósito de grasa» que acompaña con frecuencia a estos dos procesos. Esta hipótesis se ha visto reforzada por los datos publicados por Sanyal et al9, que demuestran que los pacientes con EHNA presentan una marcada resistencia periférica a la insulina, incluso aquellos con un peso corporal normal. En cualquier caso, los mecanismos por los que las alteraciones metabólicas asociadas a la resistencia a la insulina contribuyen a la aparición de esteatosis o EHNA no se conocen con exactitud, aunque se cree que la hiperinsulinemia resultante podría inducir una situación de estrés oxidativo intrahepático que, como se verá más adelante, en la actualidad se reconoce como uno de los factores más importantes en la patogenia de la EHNA.
La dislipemia, ya sea hipertrigliceridemia, hipercolesterolemia o ambas, se asocia con frecuencia a la EHNA, y se han comunicado cifras muy variables, que oscilan entre el 20 y el 81%1,2,10. En un estudio reciente en el que hemos analizado de forma prospectiva a un grupo de 46 pacientes con obesidad mórbida, encontramos que la prevalencia de dislipemia en los pacientes obesos con signos histológicos de EHNA fue del 22%6.
La cirugía abdominal derivativa para el tratamiento de la obesidad mórbida, como la gastroplastia y el bypass intestinal, también se ha descrito como factor etiológico de la EHNA10,11. El bypass intestinal, yeyunocólico o yeyunoileal es una intervención para el tratamiento de la obesidad mórbida que ha caído en desuso por las frecuentes complicaciones que origina. La mayoría de los enfermos sufren esteatosis hepática antes de la intervención, pero después un 20-40% de los casos desarrollan una EHNA, que puede evolucionar a fallo hepático fulminante en el 2-8% de éstos11. Un hecho que apoya la conexión etiológica entre la cirugía y la EHNA es que la reconstrucción del bypass suele mejorar de forma progresiva las lesiones hepáticas11.
La EHNA se ha relacionado con la ingestión de fármacos, como el maleato de perhexilina (actualmente retirado del mercado) y la amiodarona (antiarrítmico)12. En la tabla I se enumeran los fármacos asociados con más frecuencia con la EHNA, como los estrógenos sintéticos, el tamoxifeno13, los glucocorticoides, la sulfasalazina, la espironolactona, el metotrexato, el naproxeno y la oxacilina, siendo ya más raros los casos de EHNA asociados con la toma de bloqueadores de los canales del calcio, como el nifedipino y el diltiazem14.
También se exponen en la tabla I otras entidades que, aunque más raramente, se han relacionado con la aparición de EHNA como la nutrición parenteral total, la diverticulosis yeyunal con sobrecrecimiento bacteriano15, el síndrome del aceite tóxico y la exposición ocupacional a hepatotóxicos16.
Un hecho que se debe tener en cuenta es que se pueden encontrar varios factores etiológicos o causas de EHNA en un mismo paciente, aunque también se han descrito casos de EHNA sin factores etiológicos predisponentes conocidos. Así, diferentes series retrospectivas que han estudiado un limitado número de pacientes han comunicado cifras del 4617 y del 42%18 de casos de EHNA que no presentaban alguno de los factores de riesgo anteriormente mencionados. Por contra, en un reciente estudio prospectivo observacional de 1.124 pacientes adultos con transaminasas elevadas se pudo comprobar que el 97% de los pacientes con signos histológicos de EHNA eran obesos o diabéticos o dislipémicos19, lo que sugiere que si se realiza un riguroso examen clínico dirigido la mayoría de los pacientes con EHNA presentarán alguno de los factores etiológicos anteriormente mencionados.
MECANISMOS PATOGÉNICOS
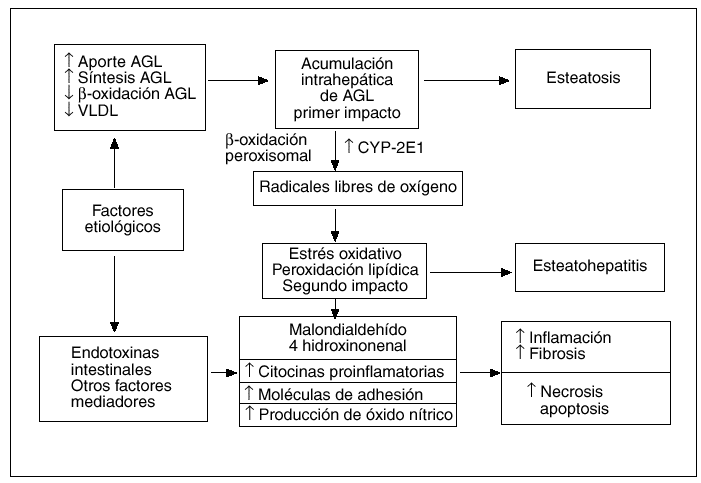
La patogenia de la EH, ya sea de etiología alcohólica o no alcohólica, no es del todo bien conocida, pero hoy día se acepta ampliamente la hipótesis de que el sustrato patológico inicial sobre el que podrán aparecer o no las lesiones inflamatorias típicas de la EH es la acumulación intrahepática de grasa en forma de triglicéridos, denominada esteatosis. La cuestión es por qué existen casos de esteatosis hepática que no evolucionan nunca hacia formas de EH20 y, por el contrario, otras progresan hacia formas evolucionadas de EH con fibrosis21. Por todo ello, parece evidente que la solución a este dilema patogénico pasa por conocer el o los mecanismos implicados en la transición de una simple esteatosis hepática, de pronóstico habitualmente benigno, a una EH con su potencial evolución a cirrosis. En los últimos años, se han acumulado ciertas evidencias experimentales, provenientes en su mayoría del estudio de modelos animales, que sugieren que al menos uno de esos nexos patogénicos entre la esteatosis y la EH puede ser la peroxidación lipídica22,23, lo que ha permitido desarrollar la teoría patogénica actualmente conocida como la «teoría del doble impacto»24 (fig. 2).
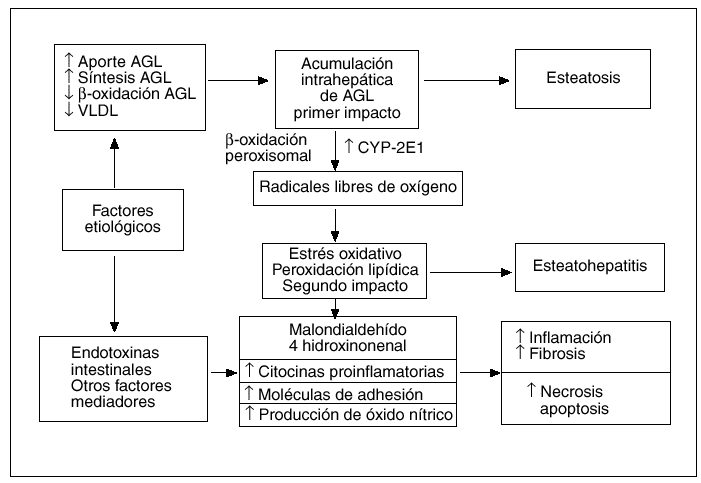
Fig. 2. Patogenia de la esteatohepatitis no alcohólica. Teoría del doble impacto.
Pessayre et al22 han comprobado que la simple esteatosis hepática en los ratones produce un cierto grado de peroxidación lipídica que no es suficiente para producir daño hepático significativo, lo que explicaría aquellos casos de esteatosis hepática que no evolucionan a EH. Para que esta apareciese, además de la esteatosis, el primer impacto, se requeriría una fuente adicional de estrés oxidativo, el segundo impacto, capaz de inducir peroxidación lipídica en un grado suficiente para vencer los mecanismos de defensa celulares, lo que pondría en marcha distintos sistemas moleculares implicados en vías proapoptóticas y proinflamatorias25.
Existen abundantes evidencias que apoyan el concepto de que la peroxidación lipídica sea uno de los mecanismos patogénicos clave de la EHNA, ya que muchas de las características histológicas de esta entidad pueden ser explicadas por el efecto de la peroxidación lipídica. La peroxidación de los lípidos de membrana puede producir necrosis celular y la formación de megamitocondrias26. Los productos finales de la peroxidación lipídica, como el 4-hidroxinonenal y el malondialdehído (MDA), pueden activar las células estrelladas que son las principales productoras de colágeno en el hígado27,28, promueven la agrupación de citoqueratinas para formar los cuerpos de Mallory22 y estimulan la quimiotaxis neutrofílica29. Por otro lado, el MDA puede contribuir a la inflamación, ya que se ha descrito que esta sustancia es capaz de activar la translocación nuclear de proteínas de la familia *B30, que actúan como factores de transcripción regulando la expresión génica de numerosas citocinas proinflamatorias y moléculas de adhesión, como el factor de necrosis tumoral alfa (TNF-*), la interleucina (IL)-8 y el ICAM-16,29.
Continuando con la teoría del doble impacto, estudios recientes han sugerido la existencia de diferentes fuentes intrahepáticas de estrés oxidativo, el denominado segundo impacto. Así, se ha descrito el aumento de expresión del citocromo P450 2E1 en modelos animales y en pacientes con EHNA32,33. La expresión de este citocromo se puede inducir, in vivo e in vitro, por la acción de los ácidos grasos y de los cuerpos cetónicos34,35. Estos datos pueden tener gran relevancia en la patogenia de la EHNA asociada a la obesidad, ya que en ella es muy frecuente encontrar valores elevados de ácidos grasos y cuerpos cetónicos en el suero y en el hígado36,37. Además de la mencionada inducción del citocromo P450 2E1, el aumento de la concentración intrahepática de ácidos grasos podría producir un incremento adicional de estrés oxidativo, induciendo vías metabólicas alternativas como la ß-oxidación de los ácidos grasos en los peroxisomas. Esta inducción tiene lugar cuando se produce una saturación de la vía metabólica fisiológica, que ocurre en las mitocondrias, por sobrecarga de sustrato o cuando esta ß-oxidación mitocondrial es inhibida por la hiperinsulinemia38,39, que con frecuencia se asocia a la obesidad40,41. A diferencia de lo que ocurre en circunstancias metabólicas normales, la desviación de la ß-oxidación de los ácidos grasos al compartimiento peroxisomal produciría peróxido de hidrógeno que, en presencia de hierro libre, generaría radicales libres altamente reactivos, como el hidroxilo. Éste podría ser el segundo impacto necesario para la progresión de esteatosis a EH asociada a la obesidad. De forma reciente se ha esbozado una novedosa teoría patogénica a raíz de la publicación de resultados obtenidos tras analizar ratones genéticamente obesos, en los que se encontraron valores más elevados de alcohol que en ratones delgados y que éstos disminuían de forma espectacular tras el tratamiento con neomicina42. Cope et al40 sugieren que en la obesidad existiría un aumento de la producción endógena de alcohol, a partir de la microflora intestinal, que podría desempeñar un papel clave en la patogenia de la EHNA asociada a la obesidad. En cualquier caso, estos interesantes datos obtenidos en modelos animales de obesidad tendrán que confirmarse en pacientes obesos, antes de considerar al alcohol endógeno como un factor con valor patogénico en seres humanos.
El papel del hierro intrahepático en la patogenia de la EHNA sigue siendo muy controvertido, y mientras que se ha publicado una serie de pacientes con EHNA en los que, en un elevado porcentaje, se ha encontrado un incremento de la concentración intrahepática de hierro en relación con estadios fibróticos avanzados43, también han salido a la luz datos de un grupo de pacientes con EHNA en los que no se observó una concentración anormalmente elevada de hierro hepático44. Más recientemente, en un estudio prospectivo de 46 pacientes con EHNA y obesidad encontramos que el 15% de ellos tenían valores séricos elevados de ferritina, pero ninguno presentaba depósitos anormales de hierro en el tejido hepático6. Por todo ello, el papel patogénico del hierro en la EHNA sigue sin estar claro, aunque se cree que la existencia de distintos patrones genéticos podrían explicar las diferencias observadas en los estudios anteriormente referidos, en los que se analizaron pacientes con EHNA que procedían de áreas geográficas muy dispares.
Otro factor que actuaría como «segundo impacto» en el modelo patogénico de la EHNA sería la endotoxina bacteriana, que estimularía la síntesis intrahepática de citocinas proinflamatorias como el TNF-*, entre otras45. Recientemente, Yang et al46 han demostrado que los ratones genéticamente obesos son más sensibles a los efectos de bajas dosis de endotoxina, desarrollando grados más intensos de EHNA que los ratones delgados. Estos autores sugieren que el mecanismo del daño hepatocelular en los ratones obesos estaría mediado por el TNF-*, aspecto que se vería apoyado en la observación de que estos ratones obesos presentan valores circulantes elevados de esta citocina proinflamatoria47. Otra evidencia experimental que apoyaría el papel patogénico de la endotoxina y del TNF-* en la EHNA asociada a obesidad es la existencia de una intensa expresión intrahepática de la enzima óxido nítrico sintasa en pacientes obesos con EHNA6, siendo un hecho bien conocido que estos dos mediadores son esenciales para que se produzca la inducción de la mencionada enzima48. Asimismo, la endotoxina podría desempeñar un papel relevante en la patogenia de la EHNA asociada a bypass intestinal, ya que en esta cirugía se produce endotoxinemia desde el asa excluida49.
Un hecho bien conocido es que no todos los pacientes con esteatosis simple evolucionan a EH, sugiriéndose que esto dependería de la existencia o no de una determinada predisposición genética. Así, se ha descrito en una entidad anatomoclínica de similares características a la EHNA, como es la EH alcohólica, la existencia de variabilidad genética en la actividad del citocromo P450 2E1 y del TNF-*50,51, relacionada con la susceptibilidad de presentar formas avanzadas de EH, lo que permite sugerir que estos polimorfismos genéticos también podrían estar implicados en la patogenia de la EHNA.
DIAGNÓSTICO
El diagnóstico de la EHNA se fundamenta en 3 criterios (tabla II):

Exclusión de un consumo excesivo de alcohol
Es un criterio básico para el diagnóstico, por lo que su exclusión se debe hacer de forma rigurosa a través de un interrogatorio exhaustivo y cuidadoso al paciente y, a menudo, a los familiares y amigos. No se podrá establecer el diagnóstico de EHNA si el paciente refiere una ingesta alcohólica segura o probable de al menos 40 g a la semana10. En ocasiones, es importante apoyarse en parámetros analíticos que pueden indicar alcoholismo activo, como el aumento del volumen corpuscular medio de los eritrocitos o la presencia de valores séricos elevados de gammaglutamil transpeptidasa (GGT)52, aunque esta última alteración analítica se puede encontrar hasta en el 28% de los pacientes con EHNA asociada a la obesidad6. En la actualidad, el parámetro analítico de mayor sensibilidad y especificidad como marcador de alcoholismo crónico es el cociente transferrina desialilada/transferrina total, cuando éste es mayor de 0,01352. En casos de difícil comunicación con el paciente y su entorno, será necesario la determinación de alcoholemia y/o alcoholuria.
Exclusión de otras causas de enfermedad hepática crónica
Se deberán realizar todos los estudios necesarios para excluir otras causas de hepatopatía crónica como las virales (virus de la hepatitis B y C), autoinmunes (hepatitis autoinmune, cirrosis biliar primaria y colangitis esclerosante primaria), metabólicas (enfermedad de Wilson, hemocromatosis y porfirias) y tóxicas (hierbas medicinales, etc.).
Lesiones histológicas típicas de esteatohepatitis
Dado que el término EH se refiere a un concepto puramente histopatológico, el diagnóstico de certeza de esta entidad sólo se puede realizar tras la demostración por biopsia hepática de las lesiones características, que son las siguientes53:
1.Depósito de grasa en el citoplasma de un número variable de células hepáticas (esteatosis), típicamente macrovesicular aunque puede ser microvesicular.
2. Focos lobulillares de degeneración-necrosis hepatocelular e inflamación en general localizados en la zona centrolobulillar (zona 3 de Rappaport), en los que predomina la degeneración balonizante y el infiltrado mixto de leucocitos polimorfonucleares y linfocitos T. Se pueden observar cuerpos hialinos de Mallory en el citoplasma de los hepatocitos degenerados. La degeneración glucogénica del núcleo de los hepatocitos o vacuolización nuclear es una lesión frecuentemente observada en la EHNA, aunque no patognomónica.
3.Fibrosis de intensidad variable que puede ser perisinusoidal, centrolobulillar o septal.
La biopsia hepática, además de ser imprescindible para el diagnóstico de la EHNA, va a aportar información clave para establecer el pronóstico evolutivo y, por tanto, para indicar el tratamiento adecuado. Con objeto de homogeneizar los criterios histopatológicos de la EHNA, tanto desde un punto de vista práctico como de investigación, se han publicado diferentes sistemas para la cuantificación de las lesiones histológicas de esta entidad, siendo actualmente el más aceptado el índice de actividad histológica propuesto por Brunt et al54, que es un sistema semicuantitativo que clasifica la actividad inflamatoria (grado) de 0 a 4 y la fibrosis (estadio) de 0 a 4 (tabla III).

Uno de los aspectos más controvertidos es determinar el momento indicado para la realización de la biopsia hepática. A modo de concepto de aplicación general, la biopsia hepática se debe realizar cuando la información esperada vaya a ser decisiva a la hora de indicar una determinada estrategia terapéutica. Como veremos después, el tratamiento de la EHNA se basa fundamentalmente en la modificación de los factores etiológicos, con el fin de evitar la progresión de la enfermedad hepática. Se sabe que la esteatosis hepática simple es una entidad habitualmente de curso benigno, por lo que para el diagnóstico de esta entidad bastaría la evidencia clínica y ecográfica pero, en cambio, se ha comprobado que la EHNA es una entidad potencialmente progresiva, y recientemente se ha observado que es una causa muy importante de cirrosis criptogénica y, por tanto, de trasplante hepático55,56. Asimismo, se ha puesto de manifiesto que existen una serie de factores que aumentan el riesgo de evolución de la EHNA hacia formas más avanzadas, incluyendo la aparición de cirrosis. Entre estos factores se han identificado la edad (> 40 años), un índice de masa corporal alto (> 40 kg/m2), un cociente GGT/GPT superior a 1 y la coexistencia de al menos dos causas de EHNA, como la diabetes mellitus y la dislipemia57,58. Por tanto, parece razonable recomendar que la biopsia hepática se realice en aquellos pacientes con sospecha de EHNA que presenten los factores de riesgo mencionados, ya que probablemente serán éstos los que puedan obtener un mayor beneficio terapéutico de la información aportada por la biopsia hepática.
TRATAMIENTO
En la actualidad, no existen medidas farmacológicas específicas para el tratamiento de la EHNA; sin embargo, dada su posible evolución progresiva y silente hacia una cirrosis parece razonable controlar los factores etiológicos que se asocian con más frecuencia a la EHNA.
Actuación sobre los factores etiológicos asociados a la esteatohepatitis no alcohólica
1. Tratamiento de la obesidad. En los pacientes obesos se deberá conseguir una reducción de peso progresiva y equilibrada mediante una dieta hipocalórica con una baja proporción en hidratos de carbono y grasas, así como con suplementos ajustados de proteínas, vitaminas y minerales, acompañándose además de un ejercicio físico programado59. El efecto beneficioso perdura a largo plazo si la pérdida de peso es mantenida; incluso se ha sugerido que la reducción gradual de peso mejora la histología hepática incluyendo la fibrosis.
2. Evitar el ayuno. El ayuno aumenta el flujo de ácidos grasos desde la periferia al hígado y deprime la síntesis de lipoproteínas, con lo que empeora la EHNA.
3. Tratamiento de la diabetes mellitus. Control de la glucemia con dieta, insulina o antidiabéticos.
4. Tratamiento de la EHNA asociada a fármacos. La única medida terapéutica ensayada ha sido suspender la administración del fármaco y, pese a ello, en algunos casos la EHNA persiste.
5. Evitar la exposición a hepatotóxicos ambientales.
6. Tratamiento de la EHNA asociada a nutrición parenteral. Se deberá reducir el aporte de glucosa sustituyéndola por lípidos. La glucosa estimula la secreción de insulina y, por tanto, inhibe la oxidación de ácidos grasos, cuya concentración aumentaría en el hígado.
7. Tratamiento de la EHNA asociada a cirugía intestinal derivativa. La reconstrucción del tránsito intestinal puede interrumpir el curso de las lesiones hepáticas, pero esto no siempre ocurre. Se ha comprobado que el metronidazol puede prevenir la aparición de EH en algunos pacientes60, posiblemente al disminuir la absorción de toxinas secundarias al sobrecrecimiento bacteriano que tiene lugar en el asa excluida.
8. Finalmente, cuando la EHNA ha evolucionado a cirrosis con hipertensión portal e insuficiencia hepática está indicado el trasplante hepático. Sin embargo, se ha constatado la aparición de recidiva de la EHNA en el hígado trasplantado. Además, se suele contraindicar para el trasplante la utilización de un hígado con marcada esteatosis, por la elevada incidencia de rechazo agudo del injerto.
Nuevas perspectivas terapéuticas
1. Ácido ursodesoxicólico (AUDC). Se ha ensayado el AUDC en el tratamiento de la EHNA basado en su acción citoprotectora sobre los hepatocitos. En un reciente ensayo clínico, en el que se comparaba la eficacia del AUDC en el tratamiento de una serie de pacientes con EHNA frente a otro grupo de pacientes tratados con un hipolipemiante (clofibrato), se demostró que este agente hipolipemiante no era eficaz en el tratamiento de la EHNA mientras que el AUDC producía una clara mejoría de la bioquímica y de la histología hepática, disminuyendo el grado de esteatosis, pero sin evidenciarse cambios significativos ni en cuanto a la inflamación ni a la fibrosis intrahepática61. No obstante, se necesitan estudios aleatorizados y controlados para evaluar el beneficio de la terapia con AUDC en los pacientes con EHNA.
2. SAMe (S-adenosil-L-metonina). Se trata de una sustancia bilógica sintetizada en el organismo, en particular en el hepatocito a partir de ATP y L-metionina. Esta molécula interviene en muchas reacciones celulares en el hígado, sobre todo de transmetilación y transulfuración, así como en la síntesis proteica y en la homeostasis lipídica. Parece que la SAMe tiene un efecto antiesteatósico y puede ser beneficiosa como tratamiento de la EHNA. Diferentes investigaciones en animales y humanos han demostrado que la SAMe es capaz de disminuir los depósitos intrahepáticos de lípidos. Este fármaco suele ser bien tolerado por los pacientes, con muy pocos efectos adversos significativos62. La SAMe ha demostrado tener un efecto beneficioso desde el punto de vista clínico, bioquímico, histológico y ecográfico sobre la EH asociada al abuso de alcohol utilizando dosis bajas (600 mg/día por vía oral o 50-100 mg/día por vía intramuscular) durante un corto período de tratamiento (uno o 2 meses). El problema es que no se dispone en nuestro país de la forma oral del fármaco, con lo cual su indicación terapéutica conllevará su administración por vía parenteral, con la consiguiente molestia para el paciente en tratamientos prolongados.
3. Colina. Los suplementos de colina se han utilizado como tratamiento de los pacientes que presentan un cuadro de EHNA asociada a nutrición parenteral en los que parece existir un déficit de colina.
4. Polimixina B oral. Se ha utilizado también en pacientes con EHNA secundaria a nutrición parenteral. Este antibiótico mejora la hepatopatía a través de disminuir la exposición del hígado a endotoxinas producidas por la flora intestinal, disminuyendo así los valores de citocinas proinflamatorias circulantes, como el TNF-*.
5.*-tocoferol. Un reciente estudio sugiere que el *-tocoferol podría mejorar la EHNA posiblemente por la modulación de las citocinas63. El *-tocoferol es un antioxidante e inhibe la expresión intrahepática de TGF-ß1 implicado en la fibrogénesis, mejorando la bioquímica hepática y también la histología.
6. Troglitazona. Se trata de un nuevo agente hipoglucemiante de la familia de las tiazolidindionas que disminuye el hiperinsulinismo a través de reducir la resistencia periférica a la insulina64, que son alteraciones metabólicas encontradas de forma muy frecuente en pacientes con EHNA y obesidad. En un ensayo piloto reciente se ha podido comprobar que la troglitazona normalizó los valores séricos de transaminasas en 4 de 6 pacientes obesos con EHNA tratados durante 2-4 meses65. No obstante, estos esperanzadores resultados no han podido ser confirmados en posteriores estudios, ya que se han comunicado varios casos de hepatotoxicidad que han hecho que este fármaco quede relegado por la rosiglitazona, con el mismo mecanismo de acción que la troglitazona pero sin presentar los efectos adversos de toxicidad hepática.
A modo de conclusión, podríamos decir que el principal objetivo de estas medidas terapéuticas es la de disminuir los depósitos intrahepáticos de grasa que constituyen la base patogénica de la EHNA, una enfermedad hepática de creciente diagnóstico en nuestro medio y que puede progresar a cirrosis, por lo que parece razonable llamar la atención sobre esta, hasta ahora considerada como «inocente», enfermedad del hígado.

