




El cáncer colorrectal (CCR) es un problema de salud pública global dada su elevada incidencia y mortalidad. Se ha estimado la aparición de más de 1,2 millones de nuevos casos y se han certificado 608.700 muertes por CCR en el año 20081. Solo en España, cada año son diagnosticados más de 25.000 nuevos casos, lo que representa el 15% del total anual de todos los diferentes tipos de tumores, y es la segunda causa de muerte por cáncer —después del de pulmón— con más de 13.000 fallecimientos anuales.
La etiología de esta enfermedad es compleja e implica una interacción de factores medioambientales y genéticos. La mayoría de los tumores (60%) tienen un origen esporádico con relación a factores ambientales2; un 27% de los casos parecen ser familiares, causados por alteraciones en genes de susceptibilidad conocidos, y el 10% de los casos son de tipo hereditario, cuyos principales ejemplos son la poliposis adenomatosa familiar (PAF) y el cáncer colorrectal hereditario no polipósico (síndrome de Lynch)3,4.
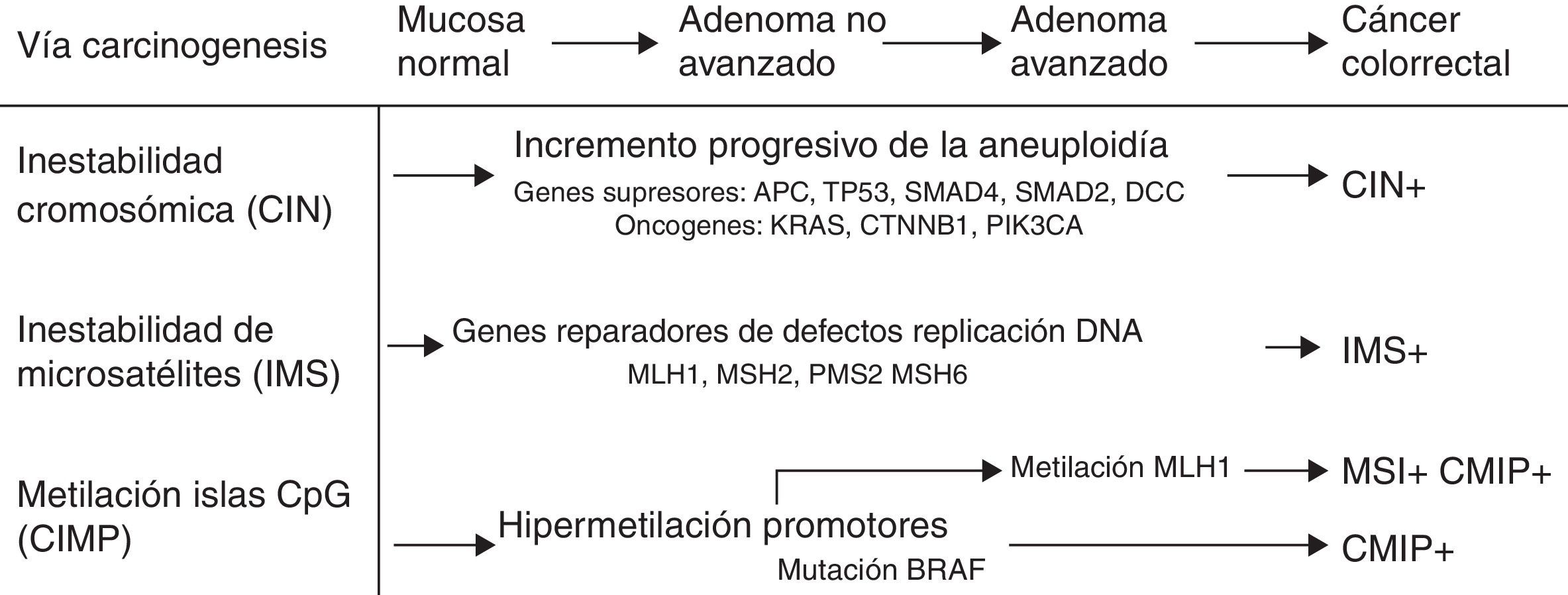
Mecanismos de carcinogénesis y genes implicadosDesde la descripción de la vía supresora en 1990 por Fearon y Vogelstein correspondiente a la secuencia adenoma-carcinoma5, se han descrito 3 vías de carcinogénesis (fig. 1). En la vía supresora o de inestabilidad cromosómica, los tumores presentan con frecuencia alteraciones del cariotipo, con ganancias y pérdidas cromosómicas, así como translocaciones. El factor presumiblemente iniciador de esta vía sería la pérdida de un gen supresor, como es el gen APC. En este grupo de CCR se ubican la mayor parte de los casos de CCR esporádicos (cerca del 85%), pero también aquellos casos de poliposis adenomatosa familiar que presentan mutaciones a nivel germinal del gen APC como base genética.

La inestabilidad de microsatélites (IMS) se produce por una inactivación heredada o adquirida en el sistema de reparación de errores de emparejamiento del ADN (dMMR). Este sistema está constituido por varios genes: MLH1, MSH2, MSH6, PMS2, entre otros. La alteración de este sistema conduce a la acumulación de alteraciones de los microsatélites, secuencias cortas y repetitivas de nucleótidos que están distribuidos a lo largo de todo el genoma6. Actualmente, se definen como tumores con IMS aquellos con alta inestabilidad o ausencia de expresión de una o más proteínas reparadoras de errores de emparejamiento del ADN en el estudio inmunohistoquímico realizado en la pieza tumoral7, y está presente en un 15% de los CCR8. La IMS se puede producir por mutaciones germinales en los genes reparadores (síndrome de Lynch) o por inactivación somática de los mismos con relación a mecanismos epigenéticos como la hipermetilación de los promotores de los genes reparadores (con mayor frecuencia a nivel de MLH1). Este mecanismo justifica los tumores esporádicos con IMS, que presentan un fenotipo clínico caracterizado por la localización en el colon derecho, un alto grado histológico y abundante infiltración linfocitaria en la pieza tumoral.
En los últimos años, se ha descrito la vía de carcinogénesis asociada a la metilación de las islas CpG (CIMP)9. Esta vía se caracteriza por la hipermetilación de la región promotora y los exones iniciales de un largo número de genes que contienen gran concentración de islas de di-nucleótidos citosina y guanina enlazados por fosfatos, produciéndose su inactivación. Esta vía se ha detectado hasta en un 35% de los pacientes con CCR10 y se denomina también vía serrada, puesto que sería la vía que justificaría la carcinogénesis de las lesiones serradas del colon11.
Por otra parte, la activación o inactivación de oncogenes o genes supresores juega un papel fundamental en el desarrollo del CCR. Así, los oncogenes Ras (HRAS, NRAS, KRAS) codifican proteínas de membrana con actividad guanosina trifosfatasa implicadas en la señalización intracelular de señales extracelulares, entre las que destacan diversos factores de crecimiento. Aproximadamente, en el 40% de los CCR se detectan mutaciones en los codones 12 y 13 del exón 2 del gen KRAS. Más recientemente, se han descrito mutaciones del gen KRAS fuera del exón 2 y también en el gen NRAS (codón 61), aunque estas son menos frecuentes. El gen BRAF constituye un miembro de la familia quinasa Raf que se encarga de regular la vía de señalización MAP quinasa/ERK. La mutación más frecuente se detecta en el codón V600E (exón 15) del oncogén BRAF y se evidencia hasta en un 30-40% de los CCR12. Estas mutaciones se asocian con frecuencia a IMS en ausencia de historia familiar de CCR13. En la mayoría de los tumores no coexisten mutaciones en KRAS y BRAF, por lo que se consideran mutuamente excluyentes14. La función del gen supresor TP53 consiste en bloquear la proliferación celular ante la presencia de ADN dañado, promueve la reparación del ADN y es capaz de dar lugar a apoptosis celular en el caso de que dicha reparación sea insuficiente. En CCR se han detectado mutaciones en TP53 hasta en un 70% de los casos15.
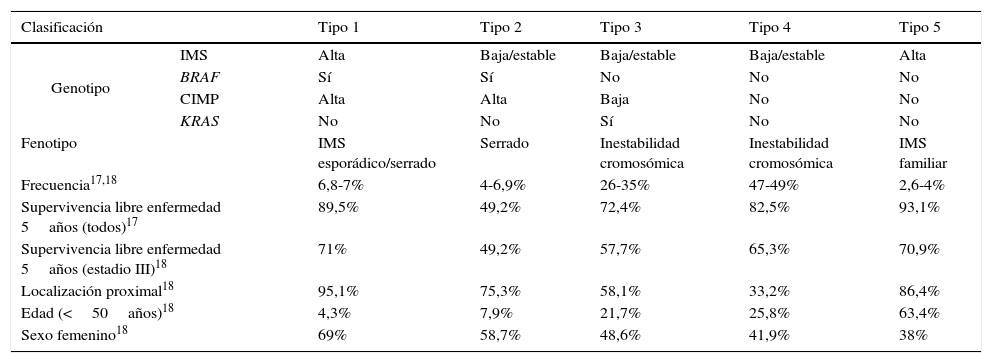
Clasificación molecular y pronósticoEn 2007, Jass16 formuló una clasificación del CCR en base al estado de IMS, al perfil metilador CIMP, a los genes supresores APC y TP53 y a los oncogenes KRAS y BRAF; en función de estas alteraciones moleculares, subclasificó al CCR en 5 tipos (tabla 1) y encontró una correlación con diversas características clinicopatológicas: lesión precursora, histología, edad al diagnóstico, sexo y ubicación. Recientemente, 2 artículos publicados han profundizado más en este tema, determinando una asociación entre esta clasificación y el pronóstico de los pacientes, tanto de forma global como en el estadio iii17,18.
Clasificación molecular del cáncer colorrectal e implicaciones pronósticas
| Clasificación | Tipo 1 | Tipo 2 | Tipo 3 | Tipo 4 | Tipo 5 | |
|---|---|---|---|---|---|---|
| Genotipo | IMS | Alta | Baja/estable | Baja/estable | Baja/estable | Alta |
| BRAF | Sí | Sí | No | No | No | |
| CIMP | Alta | Alta | Baja | No | No | |
| KRAS | No | No | Sí | No | No | |
| Fenotipo | IMS esporádico/serrado | Serrado | Inestabilidad cromosómica | Inestabilidad cromosómica | IMS familiar | |
| Frecuencia17,18 | 6,8-7% | 4-6,9% | 26-35% | 47-49% | 2,6-4% | |
| Supervivencia libre enfermedad 5años (todos)17 | 89,5% | 49,2% | 72,4% | 82,5% | 93,1% | |
| Supervivencia libre enfermedad 5años (estadio III)18 | 71% | 49,2% | 57,7% | 65,3% | 70,9% | |
| Localización proximal18 | 95,1% | 75,3% | 58,1% | 33,2% | 86,4% | |
| Edad (<50años)18 | 4,3% | 7,9% | 21,7% | 25,8% | 63,4% | |
| Sexo femenino18 | 69% | 58,7% | 48,6% | 41,9% | 38% | |
CIMP: CpG island methylator phenotype; IMS: inestabilidad de microsatélites.
El estudio de Phipps et al17 incluyó 2.706pacientes con CCR diagnosticados entre 1998 y 2007, que fueron clasificados en los 5 subtipos descritos previamente por Jass16. Así, los pacientes con perfil metilador CIMP (1 y 2) tenían una edad más avanzada al diagnóstico y solían ser mujeres. Las lesiones tumorales de los pacientes con IMS alta o perfil metilador (1, 2, 5) se ubicaron predominantemente proximales al ángulo esplénico. Por otra parte, en el tipo 2 (IMS baja, CIMP alta, mutación BRAF) se diagnosticaron en un estadio más avanzado, con una supervivencia a los 5años inferior al resto de los subtipos. En cambio, la supervivencia fue superior en los CCR con IMS alta (subtipos 1 y 5). Finalmente, la mortalidad fue significativamente inferior en el subtipo 3 (IMS baja, CIMP negativa, mutación KRAS), cuando se comparó con el subtipo 4 (IMS baja, CIMP negativa, no mutaciones) con una razón de riesgos proporcionales de 1,32.
Estos resultados fueron corroborados por el grupo de Sinicrope18 en un estudio análogo en el que se analizaron las características moleculares (mutaciones BRAF y KRAS, IMS y metilación del promotor MLH1) de 2.720 CCR en estadio III tratados con quimioterapia adyuvante (tabla 1). En este trabajo, las características clínicas de cada uno de los diferentes subtipos fueron consistentes con los resultados descritos en el estudio de Phipps et al17, tal como se muestra en la tabla 1. Los pacientes con IMS baja y con mutaciones en BRAF (Hazard Ratio [HR] 1,43; intervalo de confianza [IC] 95% 1.11-1.85) o KRAS (HR 1,48; IC 95% 1,27-1,47) tenían peor supervivencia a los 5años que aquellos sin mutaciones en dichos genes. Por otra parte, la supervivencia libre de enfermedad de los pacientes con IMS baja sin mutaciones en BRAF o KRAS es similar a la de los pacientes con IMS alta.
Cabe destacar la diferencia entre las supervivencias detectadas entre los 2 subtipos con perfil metilador (1 y 2). Aunque estos 2 subtipos son similares respecto a las características clínicas (edad avanzada, predominio de sexo femenino y ubicación proximal), la presencia de IMS alta incrementa la probabilidad de supervivencia. La mayoría de los estudios previos que habían evaluado la importancia pronóstica de IMS, CIMP y mutaciones en BRAF y KRAS habían evaluado estos marcadores de forma individual. Un metaanálisis reciente ha asociado una IMS alta a un incremento de la supervivencia global del 40% (IC 95% 31-47%)19 y la mutación BRAF V600E se ha asociado con una peor supervivencia. Por su parte, los estudios de supervivencia en relación con el perfil metilador y las mutaciones en KRAS y NRAS no se habían mostrado consistentes, probablemente porque no se habían tomado en consideración el resto de características moleculares.
La base biológica de las diferencias observadas en la supervivencia CCR subtipo específico sigue siendo un tema importante para la investigación futura. De hecho, aunque los subtipos 2 y 3 fueron diagnosticados en una etapa avanzada en el estudio de Phipps et al., los autores indican que este peor pronóstico está relacionado con una historia natural más agresiva, tal como se confirma en el estudio de Sinicrope et al.17,18.
Implicaciones terapéuticas de la clasificación molecularLos resultados de los 2 artículos son una base para establecer un valor pronóstico de la clasificación molecular en el CCR. Sin embargo, estos estudios no fueron diseñados para analizar diferencias de respuesta al tratamiento en función del subtipo. En este sentido, es ampliamente conocida la ausencia de respuesta al 5-fluorouracilo en monoterapia en pacientes con IMS alta o perfil metilador20,21. Por este motivo, en estadios intermedios (TNM II) con mejor pronóstico se debe evaluar la IMS antes de plantear la necesidad y el tipo de quimioterapia adyuvante22,23. Asimismo, el estado mutacional del gen KRAS/NRAS es un factor predictivo de respuesta a terapias específicas frente al receptor del factor de crecimiento epidérmico como el cetuximab y el panitumumab. Cabe destacar que no solo los pacientes con el gen KRAS/NRAS mutado no responden a estos tratamientos, sino que los pacientes con KRAS/NRAS nativo, pero con mutación en el gen BRAF, tienen un intervalo libre de enfermedad y una supervivencia global inferior24. Por este motivo, parece necesaria la determinación de mutaciones en KRAS/NRAS y se recomienda, también en el gen BRAF, en todo paciente con CCR metastásico para la correcta elección del tratamiento antineoplásico.
Otro avance significativo en el tratamiento del CCR ha sido publicado recientemente. La vía de muerte programada 1 (PD-1) es un sistema de retroalimentación negativo que controla la respuesta inmune citotóxica Th1. En muchos tumores, esta vía está activa, produciendo una inactivación de los linfocitos T supresores y por lo tanto la ausencia de respuesta inmune. El bloqueo con anticuerpos dirigidos produce respuestas clínicas relevantes en diversos tumores. Sin embargo, en el estudio de Topalian et al. se demostró que solo uno de 33pacientes con CCR respondía al bloqueo de PD-1, y esta respuesta se asoció a la presencia de IMS alta25. Por este motivo, Le et al. han llevado a cabo un estudio fase II para evaluar la actividad clínica de pembrolizumab en 41pacientes con cánceres metastásicos refractarios a tratamiento en función de la IMS. En este estudio, los pacientes con CCR IMS alta tratados con pembrolizumab tuvieron una mayor tasa de respuesta (40% vs. 0%), supervivencia libre de progresión (78% vs. 11%) y un menor riesgo de progresión de la enfermedad (HR 0,04; IC 95% 0,01-0,21) o muerte (HR 0,18; IC 95% 0,03-1,01) respecto a los pacientes con IMS baja. Por otra parte, los pacientes con cánceres no colorrectales con IMS alta tuvieron respuestas similares a aquellos con CCR con IMS alta26.
En conclusión, los últimos hallazgos obtenidos, junto a la información ya conocida, ponen de relieve que la clasificación molecular del CCR es relevante a la hora de establecer y comunicar el pronóstico de esta enfermedad a los pacientes. De igual forma, a la hora de decidir el curso terapéutico (tratamiento adyuvante, quimioterapia en CCR metastásico en primera línea y tras progresión) se requiere conocer las alteraciones moleculares implicadas en el desarrollo del tumor. Por lo tanto, en un futuro cercano, la toma de decisiones en los pacientes con CCR se verá determinada no solo por el estadio tumoral, sino también por los diferentes subtipos moleculares descritos en la literatura.
FinanciaciónJoaquín Cubiella ha recibido una beca de intensificación a través del programa «BIOCAPS» financiado por la Comisión Europea (FP-7-REGPOT 2012-2013-1, Grant agreement núm. FP7- 316265).

