



La determinación de las concentraciones plasmáticas de antiepilépticos para el tratamiento y profilaxis de la epilepsia es una de las estrategias que permiten mejorar los resultados clínicos, reduciendo los efectos adversos y aumentando la efectividad.
El objetivo de este artículo es revisar los aspectos básicos en la determinación de los antiepilépticos mediante un documento de consenso realizado y avalado por el grupo de trabajo de farmacocinética y farmacogenética de la Sociedad Española de Farmacia Hospitalaria (PK.gen).
Monitoring plasma levels of antiepileptic drugs for the treatment and prophylaxis of epilepsy is one of the strategies enabling clinical results to improve by reducing adverse affects and increasing effectiveness.
The objective of this article is to review the basic aspects in the monitoring of antiepileptic drugs using a consensus document prepared and endorsed by the pharmacokinetics and pharmacogenetics working group (PK.gen) of the Sociedad Española de Farmacia Hospitalaria (Spanish Society of Hospital Pharmacists).
La monitorización farmacocinética de los antiepilépticos tiene como objetivo principal la optimización del tratamiento a partir del estudio de las concentraciones del fármaco en las matrices biológicas. La individualización de la posología no es tarea fácil, debido a la presencia de factores como: a) la amplia variabilidad farmacocinética interindividual de los fármacos antiepilépticos; b) el empleo de estos fármacos como profilaxis para el control de las crisis epilépticas a largo plazo, y c) no haber definido ninguna relación entre la eficacia y algún marcador biológico que ayude a la toma de decisiones1.
Es evidente que el tratamiento de la epilepsia se ha beneficiado de la determinación de las concentraciones séricas de los tratamientos. La elevada variabilidad inter e intraindividual, la correlación entre las concentraciones séricas y el efecto, las interacciones farmacológicas, etc., aconsejan la individualización de la terapia antiepiléptica mediante su determinación farmacocinética2.
No obstante, la «popularización» de estas técnicas ha llevado, en ocasiones, al uso incorrecto de la determinación, utilizándose en situaciones injustificadas que pueden ocasionar molestias al paciente o provocando ajustes incorrectos de dosis al equiparar el intervalo óptimo de las concentraciones séricas de un fármaco con el intervalo de normalidad, lo que reduciría, en estas situaciones, la eficiencia de los tratamientos3.
El objetivo de este artículo es revisar los aspectos básicos en la determinación de los antiepilépticos mediante un documento de consenso realizado y avalado por el grupo de trabajo de farmacocinética y farmacogenética de la Sociedad Española de Farmacia Hospitalaria (PK.gen).
Consideraciones generales e intervalos terapéuticos de los distintos fármacosLos intervalos terapéuticos son intervalos de concentraciones relacionadas con la probabilidad de una determinada respuesta. En este caso, la probabilidad de que un ámbito de concentraciones séricas se asocie a una respuesta clínica en un elevado porcentaje de pacientes con una mínima incidencia de efectos adversos. La mayor parte de los estudios consideran como medida de eficacia clínica la reducción en un 50% en la frecuencia de crisis epilépticas. Por ello, los intervalos terapéuticos deben utilizarse y denominarse como intervalos de referencia. El verdadero intervalo terapéutico debería ser definido de forma individual, como el intervalo de concentraciones asociado a la mejor respuesta posible en un determinado individuo3,4.
Las recomendaciones terapéuticas nunca deben realizarse considerando únicamente las concentraciones séricas. Las manifestaciones clínicas son de vital importancia para la toma de decisiones. Es importante señalar que la dosis no debe aumentarse en aquellos pacientes que presenten un buen control de las crisis aunque las concentraciones séricas del antiepiléptico se encuentren por debajo de los límites de referencia establecidos.
En general, hay que diferenciar entre los intervalos de referencia de los antiepilépticos tradicionales (fenobarbital, fenitoína, ácido valproico, carbamacepina y primidona) y de los nuevos antiepilépticos. Existe, como es lógico, mayor evidencia, para los antiepilépticos tradicionales. Para los nuevos antiepilépticos resultan de gran utilidad los índices concentración/dosis, que informan sobre situaciones de incumplimiento terapéutico o interacciones no descritas con anterioridad1.
Los estudios de antiepilépticos realizados en niños muestran un comportamiento farmacocinético diferente respecto al observado en adultos. El aclaramiento plasmático y el volumen aparente de distribución se ven, generalmente, aumentados en la población pediátrica. Además, presentan una mayor variabilidad farmacocinética interindividual y enfermedades convulsivas más resistentes al tratamiento farmacológico.
En pacientes con insuficiencia renal o hepática que tomen antiepilépticos cuya principal vía de eliminación esté alterada será recomendable la determinación de sus concentraciones plasmáticas para conseguir tratamientos seguros y eficaces5.
Los valores de los intervalos de referencia que se utilizarán en este artículo provienen de las recomendaciones realizadas por la subcomisión de monitorización farmacoterapéutica de la Liga Internacional contra la Epilepsia4.
Fármacos antiepilépticos. Intervalos de referenciaCarbamacepinaEstudios retrospectivos y observacionales recomiendan para el tratamiento de las crisis comiciales, trastornos psiquiátricos y neuralgia del trigémino, concentraciones séricas de carbamacepina, en monoterapia, entre 4-12mg/l (17-51μmol/l). En el ajuste individual debe considerarse este intervalo variable, debido principalmente a las variaciones en la fracción libre de fármaco, a la contribución del metabolito activo (10,11-epóxido-carbamacepina) y a la propia variabilidad interindividual en la respuesta terapéutica6.
La 10,11-epóxido-carbamacepina se metaboliza mediante la enzima epóxido hidrolasa a un metabolito diol inactivo. La relación entre ambos metabolitos y entre la carbamacepina y ellos mismos proporciona una información clave en situaciones de intreracciones farmacológicas.
En pacientes en los que la carbamacepina se asocia a antiepilépticos inductores enzimáticos se puede presentar sintomatología tóxica con concentraciones de carbamacepina en el intervalo de referencia, o incluso con valores inferiores, debido al aumento en la producción de su metabolito activo4.
Igualmente, si la carbamacepina se asocia a lamotrigina, concentraciones séricas inferiores a las descritas anteriormente para monoterapia permiten mantener al paciente libre de crisis epilépticas. Tanto en este caso como en asociación con antiepilépticos inductores se recomienda no sobrepasar concentraciones séricas de carbamacepina de 8mg/l (34μmol/l), por el riesgo de aparición de efectos adversos. La administración junto a quetiapina produce en ocasiones síntomas de toxicidad con somnolencia, ataxia, etc., por inhibición de la epóxido hidrolasa y acumulación de la 10,11-epóxido-carbamacepina.
EtosuximidaEstudios retrospectivos y observacionales demuestran que la eficacia terapéutica de la etosuximida en pacientes adultos y pediatricos con crisis de ausencia se alcanza con concentraciones séricas de 40-100mg/l. Sin embargo, en pacientes con crisis resistentes o de ausencia son necesarias concentraciones séricas próximas a 150mg/l. Los pacientes con lesiones estructurales del sistema nervioso central tienen, en principio, mayor probabilidad de respuesta al tratamiento4,7.
FenobarbitalLa eficacia terapéutica de fenobarbital en pacientes adultos se ha asociado a un intervalo de concentraciones séricas bastante amplio: 10-40mg/l (43-172μmol/l). El tratamiento de las crisis parciales puede requerir concentraciones de fenobarbital más elevadas que las crisis generalizadas6,7.
FenitoínaLa eficacia terapéutica de la fenitoína en pacientes adultos se asocia a un amplio intervalo de concentraciones séricas: 10-20mg/l (40-79μmol/l), con una gran variabilidad y solapamiento de los márgenes de eficacia con los de toxicidad.
La fenitoína tiene un elevado porcentaje de unión con la albúmina sérica (90%). Las situaciones de hipoalbuminemia, como la cirrosis hepática, el embarazo o el paciente crítico, obligan a corregir la concentración total de fenitoína, pues se corresponden con una mayor proporción de fenitoína libre, que es la farmacológicamente activa6.
En neonatos se ha establecido un intervalo diferente de concentraciones totales de fenitoína, 6-14mg/l (25-55μmol/l), por su menor unión a la albúmina sérica neonatal8. Del mismo modo, en ancianos el intervalo de referencia también disminuye debido a una mayor sensibilidad a la fenitoína y a posibles alteraciones en la fracción libre de fármaco.
GabapentinaLa eficacia terapéutica de la gabapentina se ha asociado a un intervalo de concentraciones séricas amplio: 12-20mg/l (70-120 mmol/l)2, por la gran fluctuación en sus concentraciones asociada a una semivida de eliminación corta.
La gabapentina tiene una absorción dependiente de la dosis, por lo que modificaciones en la posología pueden requerir la monitorización farmacocinética. Se elimina por vía renal, siendo necesario realizar ajustes posológicos cuando la función renal se altera de forma significativa1.
La cimetidina disminuye el aclaramiento plasmático de gabapentina y los antiácidos (compuestos de aluminio y magnesio) pueden reducir su absorción más de un 24%1.
LamotriginaMorris et al9 sugieren un intervalo de concentraciones de lamotrigina para pacientes con epilepsia resistente de 3-14mg/l9. Otros autores lo amplían hasta 2,5-15mg/l (10-60μmol/l). Los pacientes tratados con una asociación de lamotrigina y vigabatrina, el intervalo de concentraciones observado para lamotrigina en pacientes respondedores correspondió a un valor de mediana de concentración plasmática de 7,9mg/l10. No obstante, hasta la fecha no se ha logrado establecer un punto de corte claro que clasifique los pacientes en respondedores o no respondedores. Cabe destacar que la incidencia de toxicidad aumenta significativamente cuando las concentraciones séricas se sitúan por encima de 15μg/ml.
La lamotrigina presenta numerosas interacciones con los inductores y los inhibidores enzimáticos. Es especialmente relevante, por su frecuencia de prescripción, la interacción con el ácido valproico que produce un aumento significativo de las concentraciones séricas de lamotrigina.
El aclaramiento plasmático de lamotrigina aumenta de forma significativa durante el embarazo. En el estudio de Petrenaite et al11 los requerimientos de dosis en el tercer trimestre de embarazo fueron el triple de los que presentaba la paciente antes de quedar embarazada.
LevetiracetamEl grupo de investigación para el cuidado de la epilepsia MINCEP12 propuso como intervalo de referencia para el levetiracetam valores entre 12-46mg/l (70-270μmol/l). El límite inferior de este intervalo fue validado por Lancelin et al13 en un estudio realizado en 2007 en 69 pacientes.
El levetiracetam presenta una eliminación predominantemente renal y por ello su aclaramiento plasmático es marcadamente dependiente de la función renal y de la edad del paciente. No obstante, una pequeña fracción de la dosis administrada de levetiracetam se elimina por metabolismo hepático y ello hace que el aclaramiento del fármaco se vea incrementado cuando se usa combinado con antiepilépticos con propiedades inductoras enzimáticas como la fenitoína o el fenobarbital4.
OxcarbamacepinaLa oxcarbamacepina es un profármaco que se metaboliza, rápida y casi totalmente, a su metabolito activo, la 10-hidroxicarbamacepina (MHD), también denominada licarbamacepina, y responsable de la acción terapéutica. Por ello, el intervalo de referencia alude a las concentraciones de este metabolito. Se ha propuesto para su monitorización farmacocinética un intervalo de concentraciones séricas de 10-hidroxicarbamacepina de 13-35mg/l (50-140μmol/l), aunque se observa una gran variabilidad y solapamiento tanto en los márgenes de eficacia como en los de toxicidad7.
La eliminación de la oxcarbamacepina y su metabolito MHD es independiente del citocromo P-450, y se produce por procesos no oxidativos como la reducción cetónica y la O-glucuronización, respectivamente. Por este motivo, su aclaramiento plasmático no está influido por la administración conjunta de otros fármacos inductores o inhibidores14. A pesar de ello se ha observado que inductores como fenobarbital, fenitoína y carbamacepina reducen la concentración del metabolito MHD.
En su monitorización rutinaria puede ser de interés el empleo de las tablas que relacionan la concentración MHD/dosis, propuesta por Armijo et al15.
PrimidonaLa eficacia terapéutica de primidona en pacientes adultos se ha asociado a un intervalo de concentraciones séricas de 5-10mg/l (23-46μmol/l). La primidona se metaboliza a fenobarbital, responsable en gran medida de su actividad, por lo que es posible individualizar el tratamiento con primidona mediante la determinación de las concentraciones de fenobarbital. La relación entre las concentraciones de primidona y fenobarbital en monoterapia es 1:2. Esta proporción aumenta en politerapia y en niños7,16.
TiagabinaEl intervalo de referencia se sitúa en valores de 20-200 ng/ml (53-532 nmol/l). La utilidad de la monitorización solo se ha demostrado en casos de sospecha de intoxicación y para confirmar la adherencia al tratamiento. En ambos casos es importante conocer el tiempo transcurrido entre la administración del fármaco y la toma de muestras debido a que su semivida biológica es muy variable, de 5-9h, en pacientes en monoterapia y de 2-4h cuando se administra con inductores enzimáticos1,17.
TopiramatoEn general, se acepta un intervalo de referencia de 5-20mg/l (15-60μmol/l). Numerosos trabajos han identificado un control epiléptico adecuado, con una buena tolerancia, con valores entre 2 y 10mg/l. Concentraciones superiores a 20mg/l se asocian a efectos adversos y encefalopatía1,3.
Ácido valproicoEl intervalo de referencia aceptado para el ácido valproico es de 50-100mg/l (347-693μmol/l). Concentraciones séricas superiores a 175mg/l se asocian con un riesgo elevado de neurotoxicidad.
Existe una escasa correlación entre la dosis administrada y las concentraciones séricas del ácido valproico debido a su elevada unión a las proteínas plasmáticas. Este proceso es saturable, incluso a concentraciones terapéuticas. En estas circunstancias, los cambios en la fracción libre del antiepiléptico no son proporcionales al aumento de la dosis. Hermida y Tutor18 proponen en pacientes con hipoalbuminemia el empleo de una tabla para corregir las concentraciones séricas de ácido valproico libre en función de los niveles de albúmina del paciente y evitar la aparición de posibles efectos tóxicos.
En ancianos es importante tener en cuenta que la fracción libre de fármaco puede estar aumentada por disminución de las proteínas plasmáticas. Sin embargo, en niños son necesarias dosis superiores a las utilizadas en adultos para alcanzar concentraciones séricas similares.
Durante el embarazo se aconseja la determinación de la concentración libre del fármaco pues se produce, debido a modificaciones de las proteínas maternas, una disminución de los valores séricos totales de ácido valproico, sin una alteración significativa de las concentraciones libres del medicamento19.
VigabatrinaLa vigabatrina se une de forma irreversible a la GABA-transaminasa, lo que provoca la falta de correlación entre la concentración y el efecto, debido al tiempo necesario para la regeneración de la enzima. No se ha podido establecer una relación adecuada concentración-efecto terapéutico. No obstante, en dosis habituales las concentraciones séricas que se alcanzan oscilan entre 0,8-36mg/l (6-279μmol/l), observándose proporcionalidad entre la dosis administrada y la concentración sérica alcanzada1,3.
ZonisamidaEl intervalo de referencia de la zonisamida se ha establecido en 10-40mg/l (47-188μmol/l). Sin embargo, en los estudios clínicos realizados la mayoría de los pacientes muestran efectos adversos con concentraciones superiores a 30mg/l1,17.
Aspectos prácticos de la monitorización farmacocinéticaSituaciones en las que se recomienda la monitorización terapéuticaLa determinación de los antiepilépticos debe realizarse solo cuando facilita la toma de decisiones farmacoterapéuticas. No está justificada la determinación sistemática.
Las indicaciones para la monitorización farmacocinética, tanto de los nuevos antiepilépticos como de los antiepilépticos tradicionales, son3,4,7,20:
- 1.
Cuando por motivos clínicos es necesario reducir el riesgo de recidivas de crisis epilépticas ajustando las concentraciones séricas del paciente dentro del considerado intervalo de referencia o en los límites superiores del mismo.
- 2.
Pacientes que reciben antiepilépticos con comportamiento farmacocinético saturable, como la fenitoína y donde es difícil predecir las concentraciones séricas a partir de la dosis inicialmente prescrita.
- 3.
Sospecha de toxicidad o cuando exista incertidumbre en el diagnóstico diferencial de los signos y los síntomas atribuibles a una toxicidad dependiente de la concentración por antiepilépticos.
- 4.
Para definir el intervalo terapéutico individual del fármaco. En pacientes con epilepsia controlada y mantenida durante un periodo de tiempo suficiente la determinación de 2 concentraciones séricas del antiepiléptico en un intervalo de varios meses son preferibles a una única determinación pues dan una estimación de la variabilidad en la medida.
- 5.
Epilepsia mal controlada. Permite identificar la causa de la recidiva o las modificaciones que deben realizarse en la pauta posológica.
- 6.
Embarazo. Durante el embarazo los cambios fisiológicos afectan a la farmacocinética de los antiepilépticos y a las concentraciones alcanzadas tanto en la mujer como en el feto. En el caso de pacientes bien controladas se recomienda determinar las concentraciones séricas del fármaco una vez cada 3 meses. En mujeres con epilepsia complicada o que toman lamotrigina u oxcarbamacepina se recomienda aumentar la frecuencia de monitorización hasta una vez al mes.
- 7.
Grupos poblacionales y estados fisiopatológicos en los que se prevé una alteración del comportamiento farmacocinético de estos fármacos, como niños y ancianos, pacientes con cirugía bariátrica, insuficiencia renal o hepática, enfermedades infecciosas, grandes quemados, pacientes críticos, etc.
- 8.
Si se producen cambios en la forma farmacéutica o en la especialidad farmacéutica.
- 9.
Sospecha de interacciones.
En general, el tiempo de extracción óptimo para la monitorización de los tratamientos antiepilépticos, administrados por vía oral, es justo antes de la dosis matinal del fármaco. La concentración obtenida se denomina concentración valle, basal, mínima o predosis.
Esta estrategia de muestreo es especialmente importante para fármacos de corta semivida biológica de eliminación. Para fármacos con semivida biológica superior a 24h, el tiempo de extracción de la muestra durante el intervalo de dosificación es menos crítico.
No obstante, cabe destacar que el momento óptimo de extracción de la muestra depende de la situación clínica y del motivo de solicitud de la determinación.
Situaciones de emergencia- –
Sospecha de intoxicación o de efecto adverso dependiente de la concentración. La toma de muestra de sangre debe practicarse lo antes posible para establecer el tratamiento más adecuado (hemoperfusión, hidratación y alcalinización, administración de carbón activo, etc.). El tiempo de muestreo con relación a la última dosis no es un factor limitante de la monitorización, aunque debe obtenerse toda la información necesaria respecto al tiempo transcurrido entre la administración del fármaco y la extracción de la muestra para realizar la correcta interpretación del resultado obtenido.
- –
Sospecha de incumplimiento terapéutico o infradosificación. Se debe realizar la extracción una vez alcanzado el equilibrio de distribución del fármaco. En caso contrario, la relación entre la concentración plasmática y la respuesta farmacodinámica puede estar alterada y puede llevar a ajustes posológicos inadecuados. En el caso de la administración de fenitoína intravenosa, la muestra posdosis debe extraerse al menos 2h después de haber finalizado la administración de la dosis.
- –
Antes de la primera dosis de la mañana, salvo fenobarbital (idealmente concentración predosis) que por su larga semivida biológica de eliminación presenta solo ligeras fluctuaciones diarias en sus concentraciones. Por otra parte, aunque la fenitoína también tiene una larga semivida biológica, el tiempo para alcanzar la concentración máxima presenta amplias oscilaciones, tanto por la forma farmacéutica como por las dosis administradas, que hacen aconsejable la determinación basal matinal.
- –
En cualquier caso, cuando se hacen mediciones comparativas es importante que los tiempos de muestreo con relación a la última dosis sean coherentes. En el caso del ácido valproico las concentraciones pueden variar hasta un 100% durante el intervalo de dosificación, y hay que tener especial cuidado en realizar siempre la extracción en el mismo momento con relación a la toma de dosis.
- –
Asimismo, se debe determinar siempre a la misma hora del día, para evitar influencias circadianas (ácido valproico y carbamacepina).
La frecuencia de las determinaciones depende de la situación clínica que originó la primera determinación y de la respuesta al tratamiento inicial.
Situaciones de emergencia- –
Tras la suspensión del tratamiento por toxicidad o efecto adverso para evaluar el riesgo de toxicidad.
- –
Transcurrido el tiempo estimado para que las concentraciones se sitúen en valores terapéuticos, considerando la semivida biológica de cada fármaco o, preferiblemente, la historia farmacocinética del paciente si se dispone de ella.
- –
A las 72-96h de iniciar el tratamiento, aunque no se haya alcanzado el estado estacionario, para descartar concentraciones subterapéuticas o tóxicas. Si tras la primera determinación se modifica la pauta posológica se realizará una nueva determinación a las 72-96h, o una vez alcanzado el estado de equilibrio estacionario.
La carbamacepina presenta un proceso autoinducción enzimática, que se inicia a las 24h de instaurar el tratamiento y que dura entre 1 y 5 semanas, por ello debe considerarse la influencia de este efecto, tanto sobre las propias concentraciones de cabamacepina como sobre la posible politerapia asociada.
Monitorización en el estado de equilibrio estacionario:- –
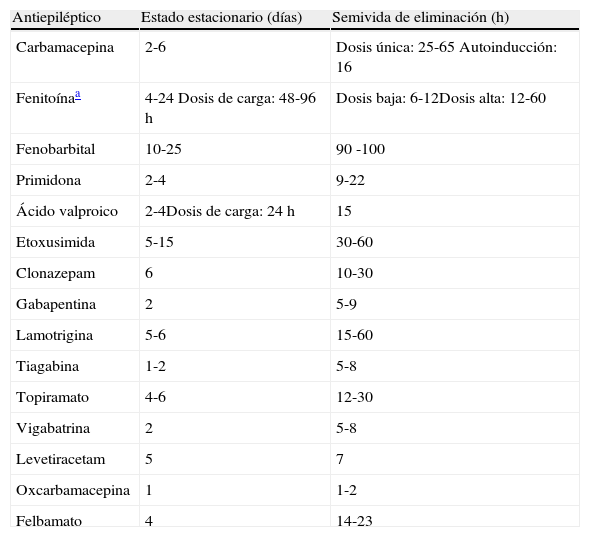
Los fármacos con cinética de eliminación de orden uno alcanzan el estado de equilibrio estacionario transcurridas, aproximadamente, cinco semividas de eliminación. La tabla 1 muestra los tiempos requeridos por los distintos antiepilépticos para alcazar el estado estacionario.
Tabla 1.Semivida de eliminación de los antiepilépticos y tiempo para alcanzar el estado de equilibrio estacionario
Antiepiléptico Estado estacionario (días) Semivida de eliminación (h) Carbamacepina 2-6 Dosis única: 25-65 Autoinducción: 16 Fenitoínaa 4-24 Dosis de carga: 48-96 h Dosis baja: 6-12Dosis alta: 12-60 Fenobarbital 10-25 90 -100 Primidona 2-4 9-22 Ácido valproico 2-4Dosis de carga: 24 h 15 Etoxusimida 5-15 30-60 Clonazepam 6 10-30 Gabapentina 2 5-9 Lamotrigina 5-6 15-60 Tiagabina 1-2 5-8 Topiramato 4-6 12-30 Vigabatrina 2 5-8 Levetiracetam 5 7 Oxcarbamacepina 1 1-2 Felbamato 4 14-23 - –
Si la cinética de eliminación es de orden cero, la semivida de eliminación aumenta cuando se alcanza el punto de saturación del metabolismo. Especialmente recomendada la monitorización cuando la respuesta al tratamiento es adecuada y las concentraciones séricas previas se encuentran en el intervalo de referencia o próximas al límite inferior.
- –
Se recomienda un control: 1) anual en pacientes adultos cumplidores, sin modificaciones en el tratamiento durante el último año y con buen control de su enfermedad; 2) semestral en pacientes pediátricos cumplidores sin modificación del tratamiento durante los últimos 6 meses y adecuado control terapéutico, y 3) trimestral o inferior cuando se producen modificaciones del tratamiento (dosis o intervalo), incorporación de medicamentos con potencial de interacción farmacológica, pacientes pediátricos no cumplidores, alteraciones en la función hepática, cardíaca o del tracto gastrointestinal, embarazo, etc.23.
La determinación sistemática de antiepilépticos con el objetivo de mantener las concentraciones en el intervalo de referencia, con independencia de la situación clínica del paciente, tiene una utilidad controvertida al aportar beneficios limitados, molestias a los pacientes y costes adicionales al sistema sanitario. Algunos autores recomiendan la monitorización sólo cuando hay una clara justificación clínica. Sin embargo, otras publicaciones recomiendan la ¿monitorización de antiepilépticos, especialmente como guía para la individualización de aquellos pacientes en los que las crisis epilépticas son poco frecuentes24. Estos mismos autores consideran además inapropiada la monitorización de pacientes en tratamiento crónico con carbamacepina o ácido valproico, cuando se producen cambios posológicos, como consecuencia de la cinética lineal predecible de estos fármacos.
Cuando se trata de pacientes ambulatorios, el primer control se realiza habitualmente 4 semanas después de administrarse la dosis total de fármaco, puesto que entonces se ha alcanzado el estado estacionario. En pacientes hospitalizados, la primera determinación también se realiza una vez alcanzado el estado de equilibrio estacionario, pero en caso administrar una dosis de choque o en situaciones urgentes la primera determinación se puede realizar antes, ajustando la posología en función de las características del paciente y extrapolando o estimando las concentraciones del fármaco en el estado estacionario.
Recomendaciones generales para la elaboración del informeEl resultado de la solicitud de monitorización debe ser un informe farmacocinético donde se recoja la siguiente información:
- –
Concentraciones séricas observadas tras la determinación analítica y su relación con el intervalo de referencia.
- –
Consecución del estado estacionario.
- –
Recomendaciones farmacoterapéuticas, incluyendo, si es necesario: dosis de carga, momento de inicio de la nueva pauta, etc.
- –
Necesidad de nuevos controles.
- –
Identificación de factores que pueden haber influido en la concentración sérica obtenida (interacciones, tiempos de muestreo inadecuados, incumplimiento terapéutico, etc.).
- –
Parámetros farmacocinéticos (opcional).
Para la elaboración del informe debe disponerse de información sobre la situación clínica del paciente, mediante comunicación con el médico responsable, el personal de enfermería y la consulta de la historia clínica. Los resultados de las concentraciones séricas y las recomendaciones efectuadas deben quedar documentados en la historia clínica del paciente, junto con la información utilizada para la realización del informe.
El informe farmacocinético, oral o escrito, debería estar disponible para el médico prescriptor antes de la administración de la siguiente dosis en pacientes hospitalizados. En pacientes externos, antes de la siguiente visita al médico responsable. Cuando se sospecha o identifican concentraciones potencialmente tóxicas o ineficaces, el tiempo máximo recomendado, tanto en pacientes ambulatorios como hospitalizados, sería de 4h.
Aspectos analíticos para la cuantificación de los antiepilépticosTipo de muestraLa matriz biológica recomendada para la cuantificación de antiepilépticos es el suero. Los anticoagulantes utilizados para la obtención de plasma pueden desplazar el fármaco de la unión a proteínas7. La heparina sódica puede activar las lipoproteín-lipasas de manera que al incrementarse las concentraciones de ácidos grasos se podrían llegar a producir desplazamientos de la unión a proteínas. Por ejemplo, el citrato u oxalato sódico disminuyen la concentración total de fenitoína y de ácido valproico. Por ello, la selección de plasma, en lugar de suero requerirá comprobar la ausencia de interacciones. Se recomienda también precaución con el uso de tubos de extracción que contengan geles pues pueden producir fenómenos de adsorción.
Técnicas analíticas para la determinación cuantitativa de antiepilépticos sometidos a monitorización clínicaEn general, las técnicas de inmunoanálisis que incluyen ensayos de polarización de fluorescencia (FPIA), ensayos inmunoenzimáticos (EMIT o CEDIA) y ensayos turbidimétricos (PETINIA, QMS) y ensayos de quimioluminiscencia (CMIA) son las más comúnmente empleadas en la práctica asistencial por su facilidad de uso y por la automatización en grandes plataformas de análisis que permiten la obtención de resultados con unos tiempos de respuesta cortos. Las técnicas cromatográficas, como la cromatografía de gases y la cromatografía líquida de alta eficacia (HPLC), son técnicas más versátiles, sensibles y específicas, circunstancias que las sitúan como una alternativa a los inmunoanálisis de gran interés. No obstante, estas técnicas cromatográficas hasta el momento presentan limitaciones; como su elevado coste y la necesidad de personal altamente cualificado. Además, suele ser necesario tratar las muestras biológicas antes de su inyección en el cromatógrafo. Así, los métodos de HPLC para antiepilépticos incluyen una extracción previa de las muestras bien a través de una precipitación, una extracción líquido-líquido o una extracción en fase sólida en cartuchos de extracción. También es habitual un proceso previo de evaporación y reconstitución del residuo obtenido.
Martinavarro-Domínguez et al25 desarrollaron un método de HPLC micelar para carbamacepina, fenobarbital y fenitoína con resultados ligeramente inferiores a los obtenidos por FPIA, aunque sin diferencias significativas para la interpretación clínica.
Dado que es muy habitual que un paciente se encuentre en tratamiento con varios antiepilépticos, su detección simultánea es interesante. Hasta el momento, no son muchos los métodos analíticos cromatográficos publicados de determinación múltiple simultánea publicados; aunque hay que destacar los trabajos de Bugamelli et al26 y Vermeij et al27 en los que se determinan simultáneamente lamotrigina, oxcarbamacepina, carbamacepina, fenobarbital, primidona, fenitoína y los metabolitos activos de carbamacepina y oxcarbamacepina.
La incorporación de los detectores de masas-masas ha revolucionado el análisis de fármacos y la cromatografía. Esto hace pensar en la cromatografía de masas como en la técnica idónea para su implantación en un futuro próximo en el laboratorio clínico; sin embargo, la necesidad de personal cualificado y su elevado coste suponen una importante limitación para su utilización en la práctica clínica asistencial. Esta es la principal razón por la que la HPLC junto con los métodos de inmunoanálisis siguen siendo las técnicas de referencia. La mayor parte de los nuevos antiepilépticos (felbamato, gabapentina, lamotrigina, levetiracetam, oxcarbamacepina, tiagabina, topiramato, vigabatrina y zonisamida) se determinan por HPLC convencional con detección ultravioleta con una importante y progresiva incorporación del detector de masas. A diferencia de la HPLC, la detección cromatográfica a través del detector de masas depende de la masa del ión molecular o del fragmento iónico del fármaco en lugar de las propiedades de absorción ultravioleta. Por ello, es una buena alternativa para la determinación de compuestos con una baja absorción en el UV. Además, dada su mayor especificidad, un fármaco puede ser fácilmente detectable a través de su ión molecular a pesar de no estar perfectamente resuelto de otros compuestos coeluyentes o interferencias. Así, el tratamiento de muestra previo a la cromatografía no es tan decisivo como en el caso de la cromatografía con detector UV. Lamotrigina, topiramato y zonisamida son 3 antiepilépticos para los cuales se han desarrollado inmunoanálisis de micropartículas (QMS) y también FPIA en el caso del topiramato. Los resultados obtenidos para las 3 técnicas de micropartículas son similares a los de HPLC.
Otras técnicas menos utilizadas son la electroforesis capilar o los electrodos de membrana selectivos. Los métodos de electroforesis capilar descritos proporcionan resultados comparables a los obtenidos con los métodos de referencia cromatográficos o inmunoanalíticos28. Gupta et al29 han desarrollado electrodos potenciométricos selectivos para la determinación de lamotrigina, felbamato y primidona y los resultados obtenidos indican que son electrodos precisos y seguros para la determinación de estos fármacos.
Tsanaclis et al30 evaluaron la precisión de los diferentes ensayos para antiepilépticos en los autoanalizadores más habituales30. La mayoría de los métodos analíticos proporcionan niveles comparables con coeficientes de variación siempre inferiores al 10% y con una exactitud en torno al 7%.
Con respecto a las técnicas cromatográficas, para todos los analitos estudiados, los resultados, en cuanto a precisión, fueron muy similares a los obtenidos con inmunoanálisis. Las pequeñas diferencias en exactitud con respecto al inmunoanálisis podrían deberse a la falta de especificidad y a la reactividad cruzada observada para algunos de los inmunoanálisis como en el FPIA de carbamacepina, cuya exactitud oscila entre el 10,7 y el 15,5%. Sin embargo, a pesar de esta reactividad cruzada debida al metabolito epóxido, con una desviación positiva, la desviación media del valor teórico es paradójicamente negativa (-8,26%). Esto parece estar relacionado con la matriz no humana utilizada en la preparación de los reactivos.
En la tabla 2 se muestran los métodos de análisis de los antiepilépticos, junto con los equipos que pueden utilizarse para su determinación.
Técnicas analíticas e instrumental empleado para la determinación de las concentraciones en fluidos biológicos de antiepilépticos
| Fármaco | Técnica analítica | Instrumental® |
| CarbamacepinaEtosuximidaFenitoínaFenobarbitalFelbamatoGabapentinaLamotriginaLevetiracetamOxcarbamacepinaPrimidonaTiababinaTopiramatoValproicoVigabatrinaZonisamida | FPIA, EMIT, CEDIA, GLC, LC-MS, CEFPIA, EMIT, CE, HPLC, GLCFPIA, EMIT, HPLC, GLC, CEDIAHPLC, GLC, LC-MS, FPIA, EMIT,CEDIA, HPLC, GLCHPLC, GC-MS, LC-MSHPLC, QMS, CEHPLC, GCHPLC, GLCFPIA, EMIT, HPLC, GLCHPLC, GC-MSFPIA, HPLC, GC, GLC-NPD, LC-MS, QMSFPIA, EMIT,CEDIA, GLC, HPLCHPLCHPLC, QMS, CE | Architect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLCTDX, ViVa E, Dimension, HPLC, GLCArchitect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCArchitect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCHPLC, GLCHPLC, GC-MS, LC-MSHPLC, Olympus AU 400, Hitachi, CDx90HPLC, GCHPLC, GCTDX, ViVa E, Dimension, HPLC, GLCHPLC, GC-MSTDX, HPLC, GC, LC-MS, Olympus AU 400, Hitachi, CDx90Architect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCHPLCHPLC, Olympus AU 400, Hitachi, CDx90 |
CE: electroforesis capilar; CEDIA: inmunoensayo de enzima donante clonada; CMIA: inmunoanálisis de quimioluminiscencia; EMIT: enzima inmunoanálisis; FPIA: inmunoanálisis de fluorescencia polarizada; GC: cromatografía de gases; GC-MS cromatografía gases-masas; GLC: cromatografía gas-líquida; GLC-NPD: cromatografía gas-líquido con detecto nitrógeno-fósforo; HPLC: cromatografía líquida de alta eficacia; LC-MS: cromatografía líquida-masas; QMS: inmunsoensayo de micropartículas.
La medición de la concentración de antiepilépticos se puede realizar también en líquido cefalorraquídeo, lágrimas y saliva, donde a excepción de la gabapentina, las concentraciones alcanzadas son similares a la fracción libre del fármaco en plasma. No obstante, la extracción de muestras de estos fluidos no resulta conveniente para la determinación sistemática por las dificultades que supone la recogida de la muestra, excepto para la saliva31.
En saliva, la evaluación de la concentración del fármaco es más compleja. La transferencia del fármaco a la saliva depende de sus propiedades físico-químicas, entre ellas el tamaño molecular, la liposolubilidad, el pKa y la unión a proteínas.
La concentración de proteínas en saliva es baja con relación a la concentración plasmática, con lo que se considera insignificante y se asume que es igual a la del agua plasmática, excepto si hay diferencias entre el pH de la saliva y el plasmático que puedan ocasionar diferencias en la concentración libre de fármaco no ionizado31.
Los fármacos antiepilépticos no ionizados en las condiciones de pH de la saliva son candidatos para la monitorización en este fluido. El efecto del pH es poco importante para fenitoína (pKa=9,2) y carbamacepina (pKa>12) que se encuentran poco afectados por cambios en el pH como consecuencia de la administración de estimulantes de la salivación. En el caso del fenobarbital (pKa=7,2) se ha propuesto la corrección de la determinación en saliva por el valor de pH para predecir más adecuadamente la concentración plasmática de este fármaco mediante la ecuación de Henderson-Hasselbach22. Otros autores han encontrado excelentes correlaciones entre saliva y concentración plasmática sin necesidad de corregir por el pH de la saliva32.
Bajo condiciones estandarizadas y controladas la saliva se puede utilizar como una matriz alternativa para la determinación de carbamacepina, fenitoína, primidona y etosuximida, puesto que se ha demostrado una buena correlación con la fracción libre de estos fármacos en plasma32. Por el contrario, se han obtenido resultados controvertidos con ácido valproico y fenobarbital33,34.
Determinación de la fracción libreHabitualmente, no es necesaria la determinación sistemática de la fracción libre de los antiepilépticos. Los fármacos que por su utilidad clínica son candidatos para la determinación de la fracción libre son aquellos con una fracción de fármaco unido a proteínas plasmáticas superior al 80%35. En estos casos, una disminución relativamente pequeña de la concentración de proteína disponible para la unión al fármaco puede tener un impacto significativo en una elevación de la concentración de fármaco libre. Igualmente, la fracción libre puede verse modificada cuando la extensión de la unión a proteínas se modifica como consecuencia de cambios en la concentración del fármaco, disponibilidad o competición y afinidad por los sitios de unión.
Los antiepilépticos que se consideran candidatos para la determinación de sus concentraciones libres son la fenitoína, el ácido valproico y la carbamacepina, a pesar de que el grado de unión de este último no alcanza el 80%. La tiagabina cumple estos criterios, aunque son necesarios más estudios para establecer su intervalo de referencia.
La utilidad clínica de la monitorización de la concentración libre de los antiepilépticos ha sido ampliamente documentada en la bibliografía. No obstante, existen resultados donde la relación entre la fracción libre y el efecto farmacológico son contradictorios35. Se ha observado una elevada variabilidad interindividual en la fracción libre de estos fármacos en pacientes con insuficiencia renal y enfermedad hepática grave. Los pacientes pediátricos con desnutrición y los neonatos, especialmente aquellos con hiperbilirrubinemia, presentan una disminución en la unión de los fármacos a las proteínas.
El ácido valproico se caracteriza por una elevada unión a proteínas plasmáticas, fundamentalmente a la albúmina, de carácter saturable a concentraciones terapéuticas (75mg/l). En aquellas situaciones en las que su aclaramiento plasmático intrínseco no se modifique, pero se altere la unión a las proteínas plasmáticas es recomendable la determinación de la fracción libre. El porcentaje de fracción libre de ácido valproico puede aumentar desde el 5%, con concentraciones totales de 10-60mg/l, al 10% cuando la concentración es de 75-100mg/l, hasta el 20-30% a concentraciones totales de 145-160mg/l, habiéndose observado incrementos en el porcentaje de fracción libre superiores al 50%18.
El ácido valproico presenta una baja tasa de extracción renal, con un aclaramiento plasmático total dependiente de su fracción libre y del aclaramiento hepático intrínseco. Una reducción en la unión a la albúmina aumentará la fracción libre, farmacológicamente activa, pero también la fracción libre disponible para ser eliminada, con lo que también aumenta, en global, el aclaramiento total de ácido valproico. Esto lleva a una disminución de la concentración plasmática total, mientras que la concentración de ácido valproico libre se mantiene o incluso aumenta. Un incremento de la dosis provocaría un aumento en la concentración no unida a proteínas, aumentando el riesgo de toxicidad. Es por ello importante detectar las situaciones en las que se produzca un desplazamiento de la unión a proteínas plasmáticas o una reducción de las mismas, como son la insuficiencia renal o hepática, hiperbilirrubinemia, exceso de ácidos grasos libres plasmáticos, uremia, interacciones farmacológicas, etc.36,37.
Tal es el caso del ácido salicílico que compite con el ácido valproico por los sitios de unión a la albúmina. Estudios in vitro han identificado incrementos de la fracción libre de ácido valproico en pacientes seropositivos al virus de la inmunodeficiencia adquirida (VIH+) como consecuencia de la hipoalbuminemia y de la posible interacción entre el ácido valproico y el tratamiento antirrretroviral35. También se recomienda determinar la fracción libre de ácido valproico en los pacientes que no están bien controlados después de un aumento de la dosis38.
Algunos autores han desarrollado métodos indirectos para la estimación de la fracción libre (KODAMA). Hermida et al13 han propuesto un modelo matemático para normalizar la concentración total de ácido valproico en pacientes con hipoalbuminemia a partir de la siguiente expresión: CN=αH·CH/6,5, donde CN es la concentración total de ácido valproico normalizada, αH es el porcentaje de fracción libre calculado para el paciente, o bien, en su defecto el presentado por estos autores para diferentes valores de albúmina sérica, y·CH es la concentración total de ácido valproico determinada. Esta metodología puede ser de utilidad para la individualización posológica en pacientes con hipoalbuminemia cuando la fracción libre del fármaco no está disponible18.
No obstante, es recomendable la determinación directa de la fracción libre. Para ello basta con separar, mediante ultrafiltración, la fracción libre de la fracción unida a proteínas plasmáticas y determinar en el ultrafiltrado la concentración de antiepiléptico39.
La fenitoína se une en un 90% a las proteínas plasmáticas, principalmente a la albúmina, pero a diferencia del ácido valproico, esta unión no es saturable. El intervalo de referencia se situa entre 0,8-2,1mg/l.
La situaciones que afectan el porcentaje de unión de la fenitoína a las proteínas plasmáticas son similares a las del ácido valproico. La unión de la fenitoína a la albúmina puede verse afectada significativamente en situaciones de uremia elevada y en situaciones que conlleven modificaciones estructurales en los sitios de unión de la albúmina. En hipercolesterolemia e hiperlipidemias mixtas, el aumento de ácidos grasos libres desplaza a la fenitoína de su unión a la albúmina observándose un incremento de la fracción libre. En pacientes con eclampsia, frecuentemente la fracción libre de fenitoína está anormalmente elevada; sin embargo, en estas situaciones ni la concentración total ni la libre son un buen predictor de la respuesta en el paciente. La unión de fenitoína a las proteínas también puede verse afectada por la presencia de otros fármacos ácidos como son ácido valproico, salicilatos, fenibutazona y sulfonilureas. Finalmente, se ha observado que en pacientes infectados por el VIH la concentración de fenitoína libre se encuentra aumentada por hipoalbuminemia o como resultado de la interacción de la fenitoína con los múltiples tratamientos que reciben estos pacientes35.
En un estudio realizado en 139 pacientes, la fracción libre de fenitoína oscilaba entre el 6,8 y el 35,3% de la concentración plasmática total. Las situaciones clínicas responsables de esta variabilidad fueron la hipoalbuminemia, interacciones farmacológicas, uremia, embarazo y grupos de edad extremos. Los autores concluyeron que la determinación de la concentración plasmática total de fenitoína era menos fiable que la concentración de fenitoína libre indicador subrogado de efectividad. Recientemente, Iwamoto et al40 reiteraron la necesidad de monitorizar la fracción libre de fenitoína en pacientes en monoterapia.
En situaciones de hipoalbuminemia, cuando no se dispone de la determinación analítica de la fracción libre, similar al procedimiento seguido con el ácido valproico, la concentración plasmática total de fenitoína se puede normalizar o corregir a partir de la ecuación de Sheiner-Tozer:
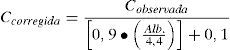
Donde, Cobservada es la concentración total de fenitoína observada tras la determinación analítica y Alb. es la concentración de albúmina del paciente expresada en g/dl38.
Del mismo modo, en pacientes con hipoalbuminemia sometidos a técnicas de depuración extrarrenal se puede modificar la ecuación anterior para normalizar la concentración plasmática total de fenitoína cambiando el valor de 0,9 por 0,43.
La carbamacepina se une en un 70-80% a las proteínas plasmáticas y su principal metabolito activo, la carbamacepina 10,11-epóxido, en un 50%. La carbamacepina presenta una menor variabilidad en la unión a proteínas plasmáticas que el ácido valproico o la fenitoína. No obstante, la utilidad de la determinación de la fracción libre de carbamacepina en los pacientes en monoterapia es menor pues se ha observado un mayor solapamiento entre las concentraciones potencialmente terapéuticas y tóxicas de carbamacepina libre (intervalo de referencia para la fracción libre de carbamacepina: 0,9-2,8mg/l)34,35. Sin embargo, en pacientes con uremia, enfermedad hepática o mujeres embarazadas la elevación inesperada de la fracción libre de carbamacepina justifica su monitorización en estos grupos poblacionales.
En resumen, la monitorización de la fracción libre de ácido valproico, fenitoína y carbamacepina está indicada en las siguientes situaciones35,36.
- –
Concentraciones séricas totales en el intervalo de referencia y presencia de signos o síntomas de toxicidad en el paciente o sin respuesta adecuada.
- –
Pacientes urémicos o con insuficiencia renal grave.
- –
Pacientes con enfermedad hepática.
- –
Pacientes con hipoalbuminemia o concentración de albúmina < 2,5g/dl.
- –
Medicación concomitante con alta unión a proteínas plasmáticas que pueda competir por la unión a proteínas plasmáticas del antiepiléptico.
- –
Pacientes con hiperlipidemias.
- –
Pacientes infectados por el VIH.
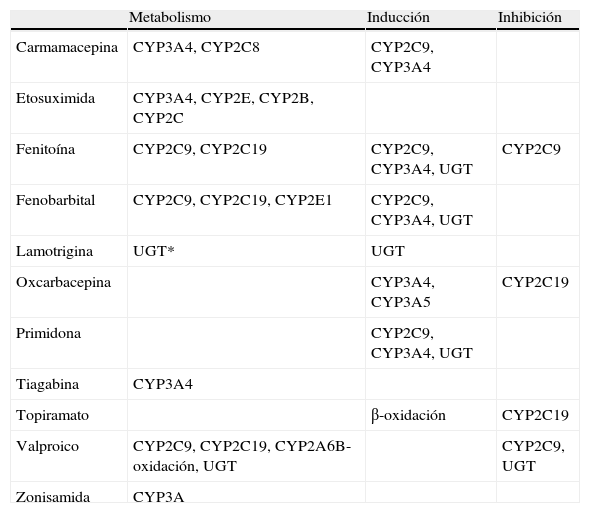
Los antiepilépticos tradicionales suelen eliminarse mediante metabolización a través del citocromo P-450 y distintos transportadores. Por ello, son objeto de interacciones medicamentosas, tanto entre ellos como con el resto de la medicación que recibe el paciente41–43. En la tabla 3 se muestran las distintas isoformas del citocromo P-450 que son responsables del metabolismo de los antiepilépticos tradicionales así como aquellas isoformas enzimáticas que pueden ser inducidas o inhibidas por estos medicamentos, provocando alteraciones en las concentraciones séricas del antiepiléptico. Las tablas 4–6 recogen las principales interacciones con relevancia clínica.
Enzimas del citocromo P-450 implicados en el metabolismo de los antiepilépticos y efecto inductor o inhibidor del fármaco sobre el citocromo P-450
| Metabolismo | Inducción | Inhibición | |
| Carmamacepina | CYP3A4, CYP2C8 | CYP2C9, CYP3A4 | |
| Etosuximida | CYP3A4, CYP2E, CYP2B, CYP2C | ||
| Fenitoína | CYP2C9, CYP2C19 | CYP2C9, CYP3A4, UGT | CYP2C9 |
| Fenobarbital | CYP2C9, CYP2C19, CYP2E1 | CYP2C9, CYP3A4, UGT | |
| Lamotrigina | UGT* | UGT | |
| Oxcarbacepina | CYP3A4, CYP3A5 | CYP2C19 | |
| Primidona | CYP2C9, CYP3A4, UGT | ||
| Tiagabina | CYP3A4 | ||
| Topiramato | β-oxidación | CYP2C19 | |
| Valproico | CYP2C9, CYP2C19, CYP2A6B-oxidación, UGT | CYP2C9, UGT | |
| Zonisamida | CYP3A |
aUGT: uridín-difosfato-glucuroniltransferasa.
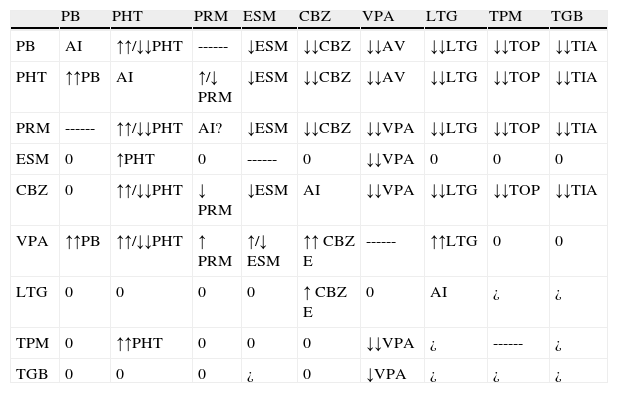
Interacciones entre antiepilépticos. Efecto sobre la concentración plasmática
| PB | PHT | PRM | ESM | CBZ | VPA | LTG | TPM | TGB | |
| PB | AI | ↑↑/↓↓PHT | ------ | ↓ESM | ↓↓CBZ | ↓↓AV | ↓↓LTG | ↓↓TOP | ↓↓TIA |
| PHT | ↑↑PB | AI | ↑/↓ PRM | ↓ESM | ↓↓CBZ | ↓↓AV | ↓↓LTG | ↓↓TOP | ↓↓TIA |
| PRM | ------ | ↑↑/↓↓PHT | AI? | ↓ESM | ↓↓CBZ | ↓↓VPA | ↓↓LTG | ↓↓TOP | ↓↓TIA |
| ESM | 0 | ↑PHT | 0 | ------ | 0 | ↓↓VPA | 0 | 0 | 0 |
| CBZ | 0 | ↑↑/↓↓PHT | ↓ PRM | ↓ESM | AI | ↓↓VPA | ↓↓LTG | ↓↓TOP | ↓↓TIA |
| VPA | ↑↑PB | ↑↑/↓↓PHT | ↑ PRM | ↑/↓ ESM | ↑↑ CBZ E | ------ | ↑↑LTG | 0 | 0 |
| LTG | 0 | 0 | 0 | 0 | ↑ CBZ E | 0 | AI | ¿ | ¿ |
| TPM | 0 | ↑↑PHT | 0 | 0 | 0 | ↓↓VPA | ¿ | ------ | ¿ |
| TGB | 0 | 0 | 0 | ¿ | 0 | ↓VPA | ¿ | ¿ | ¿ |
¿: interacción no conocida o investigada; ↑: puede aumentar ligeramente la concentración plasmática; ↑↑: puede aumentar intensamente la concentración plasmática; ↓: puede disminuir ligeramente la concentración plasmática; ↓↓: puede disminuir intensamente la concentración plasmática; 0: sin interacción; AI: autoinducción; CBZ E: carbamacepina epóxido; CBZ: carbamacepina; ESM: etosuximida; LTG: lamotrigina; PB: fenobarbital; PHT: fenitoína; PRM: primidona; TGB: tiagabina; TPM: topiramato; VPA: ácido valproico.
Efecto inductor o inhibidor de los antiepilépticos sobre otros fármacos
| Antiepiléptico | Fármaco |
| Inductores enzimáticos | |
| • Fenitoína• Fenobarbital• Carbamacepina• Primidona• Oxcarbamacepina | Antibióticos: praziquantel, albendazol, cloranfenicola, (sólopb), doxiclina, metronidazolAntifúngicos: griseofulvina, itraconazolAntivirales: nevirapina, efavirenz, delavirdina, indinavir, ritonavir, saquinavirAntineoplásicos: ciclofosfamida, ifosfamida, busulfán, tenipósido, etopósido, paclitaxel, metrotrexato, alcaloides de la vincaAntiarrítmicos: disopiramidab, mexiletina, quinidina, amiodaronabAntihipertensivos: propranolol, metoprolol, alprenolol, nifedipino, felodipino, nimodipino, nisoldipino, verapamilo, losartáncCardiotónicos: digoxinadHipocolesterolemiantes: atorvastatina, lovastatina, simvastatina, fluvastatinaAnticoagulantes orales: dicumarol, warfarinaInmunosupresores: ciclosporina, tacrolimus, sirolimusAntidepresivos: amitriptilina, nortriptilina, imipramina, desimipramina, clomipramina, doxepina, mianserina, somifensina, bupropiónf, nefazodonam, citalopram, paroxetinaAntipsicóticos: haloperidol, clorpromazina, tioridazina, clozapina, olanzapina, risperidona, quetiapina, ziprasidonaBenzodiacepinas: clobazam, diazepam, nordiazepam, clonazepam, alprazolam, midazolamgCorticoides: hidrocortisona, cortisol, dexametasona, prednisona, prednisolona, metilprednisolonaAnticonceptivos oraleshOtros: teofilina, fentanilo, metadona, meperidina, paracetamol, tiroxina, y bloqueantes neuromusculares |
| Inhibidores enzimáticos | |
| • Ácido valproico | Antihipertensivos: nimodipinoCitostáticos: nitrosourea-cisplatino1Antivirales: nevirapina, efavirenz, delavirdina, indinavir, ritonavir, saquinavir, zidovudinaHipocolesterolemiantes2: atorvastatina, lovastatina, simvastatina, fluvastatinaInmunosupresores: ciclosporina3Antidepresivos triciclicos: amitriptilina, nortriptilina, clomipramina, paroxetinaAntipsicóticos: haloperidol, clorpromazina, tioridazina, olanzapina, quetiapina, ziprasidona, diazepam, lorazepam |
La formación de metabolitos activos de la disopiramida y amiodarona hacen más impredecible el efecto de esta interacción. La magnitud de la interacción con las dihidropirimidinas antagonistas del calcio no aconsejan su uso con inductores enzimáticos.
La fenitoína no afecta las concentraciones plasmáticas de losartán, pero reducen las de su metabolismo activo.
Sólo esta descrita, como interacción significativa, con fenitoína.
eInteracción compleja. Monitorizar el INR.
La significación clínica con bupropión no está bien definida por la existencia de un metabolito activo.
Poca relevancia, clínica debido al amplio intervalo terapéutico de las benzodiacepinas, excepto para el midazolam por vía oral.
No afectan el metabolismo de los anticonceptivos orales la gabapentina, lamotrigina, levetiracetam, tiagabina, zonisamida, vigabatrina y ácido valproico, tampoco el topiramato a dosis de 200mg; 1riesgo de toxicidad hematológica; 2no existen datos concluyentes. No obstante, se recomienda precaución por la toxicidad potencial de la interacción; 3no afecta la concentración. Riesgo de hepatotoxicidad.
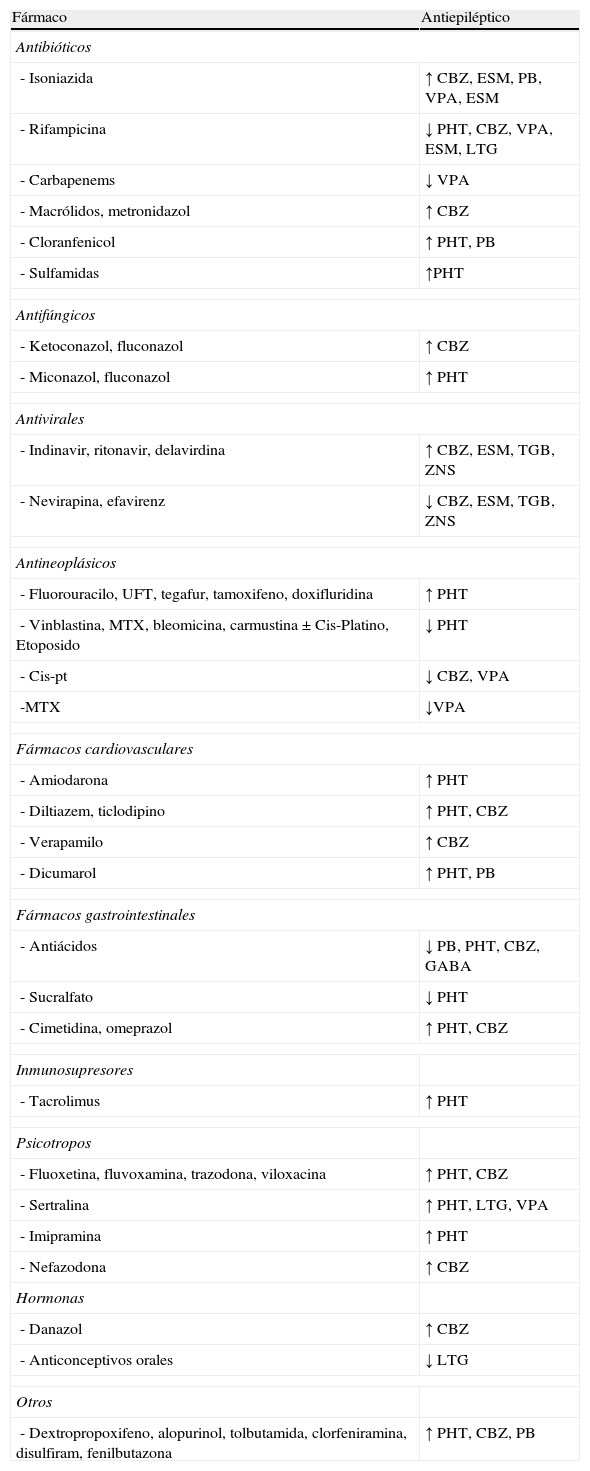
Fármacos que aumentan (↑) o disminuyen (↓) la concentraciones sérica de los antiepilépticos
| Fármaco | Antiepiléptico |
| Antibióticos | |
| - Isoniazida | ↑ CBZ, ESM, PB, VPA, ESM |
| - Rifampicina | ↓ PHT, CBZ, VPA, ESM, LTG |
| - Carbapenems | ↓ VPA |
| - Macrólidos, metronidazol | ↑ CBZ |
| - Cloranfenicol | ↑ PHT, PB |
| - Sulfamidas | ↑PHT |
| Antifúngicos | |
| - Ketoconazol, fluconazol | ↑ CBZ |
| - Miconazol, fluconazol | ↑ PHT |
| Antivirales | |
| - Indinavir, ritonavir, delavirdina | ↑ CBZ, ESM, TGB, ZNS |
| - Nevirapina, efavirenz | ↓ CBZ, ESM, TGB, ZNS |
| Antineoplásicos | |
| - Fluorouracilo, UFT, tegafur, tamoxifeno, doxifluridina | ↑ PHT |
| - Vinblastina, MTX, bleomicina, carmustina±Cis-Platino, Etoposido | ↓ PHT |
| - Cis-pt | ↓ CBZ, VPA |
| -MTX | ↓VPA |
| Fármacos cardiovasculares | |
| - Amiodarona | ↑ PHT |
| - Diltiazem, ticlodipino | ↑ PHT, CBZ |
| - Verapamilo | ↑ CBZ |
| - Dicumarol | ↑ PHT, PB |
| Fármacos gastrointestinales | |
| - Antiácidos | ↓ PB, PHT, CBZ, GABA |
| - Sucralfato | ↓ PHT |
| - Cimetidina, omeprazol | ↑ PHT, CBZ |
| Inmunosupresores | |
| - Tacrolimus | ↑ PHT |
| Psicotropos | |
| - Fluoxetina, fluvoxamina, trazodona, viloxacina | ↑ PHT, CBZ |
| - Sertralina | ↑ PHT, LTG, VPA |
| - Imipramina | ↑ PHT |
| - Nefazodona | ↑ CBZ |
| Hormonas | |
| - Danazol | ↑ CBZ |
| - Anticonceptivos orales | ↓ LTG |
| Otros | |
| - Dextropropoxifeno, alopurinol, tolbutamida, clorfeniramina, disulfiram, fenilbutazona | ↑ PHT, CBZ, PB |
CBZ: carbamacepina; ESM: etosuximida; GABA: gabapentina; LTG: lamotrigina; PB: fenobarbital; PHT: fenitoína; PRM: primidona; TGB: tiagabina; TPM: topiramato; VPA: valproico ácido; ZNS: zonisamida.
En la actualidad existe un enorme interés en determinar la influencia de los polimorfismos genéticos sobre la farmacocinética con objeto de optimizar la farmacoterapia. Sin embargo, los datos sobre la relevancia clínica en la disposición de los fármacos son conflictivos. Williams et al44 evaluaron los motivos de estas discrepancias, concluyendo que la mayor parte de los estudios no presentaron el tamaño muestral necesario para que la magnitud del efecto medido reflejara el efecto real del polimorfismo.
Entre las distintas isoformas enzimáticas que intervienen en el metabolismo de los antiepilépticos y que poseen variantes alélicas de probada y significativa influencia en su disposición, hay que destacar la CYP2C9 y la CYP2C19. Las variantes alélicas de interés para estas 2 isoformas son *2 y *3, al menos en la población caucásica. El trabajo de Klotz analiza estos aspectos44.
El PB es metabolizado parcialmente por el CYP2C19, pero el limitado porcentaje de fármaco eliminado por esta vía hace que la repercusión de los distintos polimorfismos de esta isoforma no tenga un impacto significativo en el manejo clínico del antiepiléptico.
En el metabolismo de la fenitoína intervienen ambas isoformas, más predominante la CYP2C9. El papel del CYP2C19 es más relevante con concentraciones elevadas. Existe falta de consenso sobre la influencia de los polimorfismos en la farmacocinética de la fenitoína, no solo por las limitaciones en el diseño de los estudios, ya mencionadas, sino además por la gran variabilidad entre grupos étnicos. Klotz describió el caso de una paciente afroamericana que mostró una variante alélica (CYP2C9*6) de muy baja frecuencia en su etnia (0,6%) y nula en los caucásicos, y presentó una exposición a fenitoína 5,8 veces superior a los poseedores de las variantes salvaje44.
En otro estudio se observó que los pacientes poseedores de la variante CYP2C9*3 podían requerir un 13% menos de dosis, siendo la única variante con repercusión clínica. Por ello, deberían valorarse conjuntamente ambas variantes genéticas para obtener resultados más ajustados. No obstante, la monitorización farmacocinética de la fenitoína y la estimación de los parámetros farmacocinéticos individualizados ya incorpora la variabilidad farmacogenética y permite diseñar pautas farmacoterapéuticas seguras y eficaces.
Las principales vías de eliminación del ácido valproico son la glucuronización, la beta-oxidación y la omega-oxidación. La isoforma CYP2C9 interviene en la formación de un metabolito denominado 4-N-valproico que está relacionado con la hepatotoxicidad producida por el antiepiléptico. Sin embargo, hasta la fecha los estudios realizados han fracasado al intentar demostrar una asociación entre las posibles variantes genéticas del CYP2C9 y la incidencia de hepatotoxicidad, posiblemente porque intervenga algún otro mecanismo independiente del CYP.
ConclusionesLa determinación de las concentraciones séricas de los antiepilepticos tiene un papel importante en la optimización del tratamiento de la epilepsia, debido principalmente a la gran variabilidad farmacocinética que presentan estos fármacos, su uso clínico en tratamientos profilácticos y su estrecho margen terapéutico. En este artículo se revisan las principales recomendaciones y aspectos prácticos para realizar una adecuada monitorización de los mismos. Los valores de referencia indicados definen las concentraciones séricas en las cuales la mayoría de los pacientes obtienen una respuesta óptima y provienen de las recomendaciones realizadas por la subcomisión de monitorización farmacoterapéutica de la Liga Internacional contra la Epilepsia. No obstante, la relación concentración/respuesta no siempre es estable en el tiempo para un mismo paciente, ya que cambios en su estado, enfermedades concomitantes, cambios en la unión a proteínas, interacciones con otros fármacos, etc., pueden modificar esta relación, por ello la monitorización de las concentraciones séricas de los antiepilépticos proporciona una importante información para el ajuste de la dosis.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

