




Los tumores neuroendocrinos de páncreas (TNEp) funcionantes representan el 1% de las neoplasias pancreáticas. Aún más infrecuentes son los productores de ACTH, que representan el 15% de las causas de secreción ectópica de ACTH1-3, tienen un comportamiento agresivo y mayoritariamente (94%) están bien diferenciados1. Cuando la opción quirúrgica no es posible, se dispone de pocas estrategias terapéuticas eficaces antitumorales frente a ellos4, siendo un reto su manejo terapéutico. Aportamos nuestra experiencia con un caso de un TNEp inicialmente no funcionante que desarrolló síndrome de Cushing (SC) a los 3años del diagnóstico y un segundo caso que comenzó con SC florido.
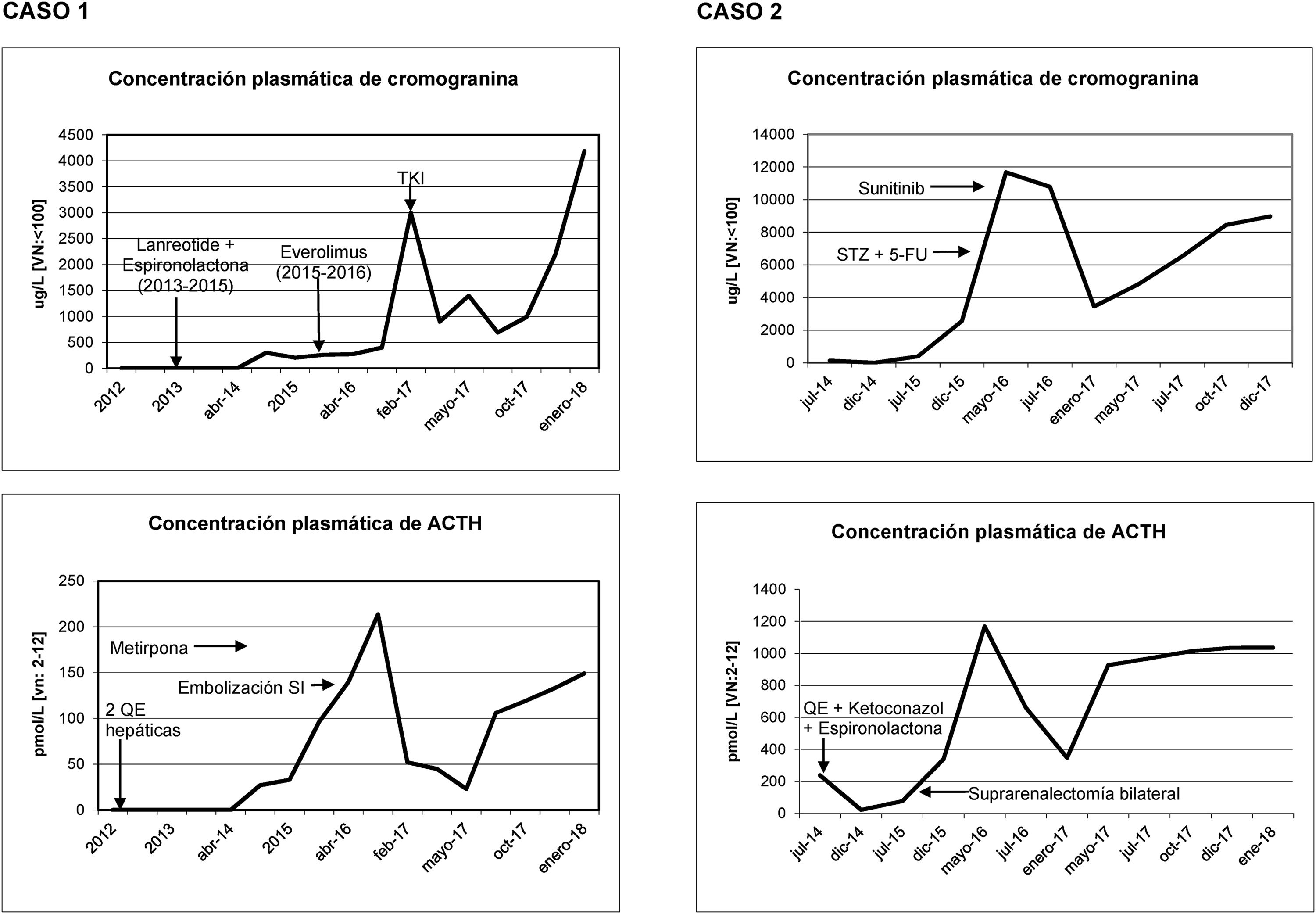
Caso 1Varón de 57 años diagnosticado en 2011 de TNEp de 2cm en la cola bien diferenciado (pT2N0, grado2 con Ki67 del 15%), intervenido mediante pancreatectomía subtotal y esplenectomía, sin signos de SC. En diciembre de 2012 presentó recidiva local y metástasis hepáticas. En junio del 2013 se realizó enucleación de la cabeza pancreática y radiofrecuencia o alcoholización hepática según el tipo de lesión. Sin embargo, por progresión hepática, en octubre de 2013 se inició lanreótida (120mg/28días) junto con espironolactona por tendencia a la hipertensión (50mg/día). En octubre de 2014, por progresión hepática, se cambió de lanreótida a everolimus 10mg/día, recibido de febrero de 2015 a marzo de 2016 y con respuesta parcial. Paralelamente, en noviembre de 2014 desarrolló SC paraneoplásico con deterioro del estado general, astenia, miopatía, anasarca, diabetes mellitus, crisis hipertensiva, tromboembolismo pulmonar bilateral e hipopotasemia grave de 2,19mmol/l con alcalosis metabólica. A la exploración física destacaba rubicundez facial e hiperpigmentación. Se detectó concentración plasmática de ACTH de 122pg/ml [valores normales (VN): 10-60], cortisol sérico de 29,5μg/dl [VN: 10-25] y cortisol libre urinario (CLU) de 143,8μg/día [VN: 20-100]. La tomografía computarizada (TC) mostró hiperplasia suprarrenal bilateral. Ante ello, se inició ketoconazol que por hepatitis tóxica se sustituyó a metirapona (2.000 mg/día hasta 4.000mg/día). Por mal control del hipercortisolismo se practicó embolización únicamente de la suprarrenal izquierda por problemas en el acceso vascular de la derecha en septiembre de 2016, persistiendo posteriormente ACTH (970pg/ml) y cortisolemia (63,3μg/dl) elevadas. Por otro lado, por progresión metastásica hepática en abril y julio de 2016 se indicó quimioembolización (QE) en dos sesiones. Radiológicamente continuaron los signos de progresión hepática y apareció lesión lítica metastásica en L1. Se inició un inhibidor de la tirosín-quinasa (Lenvatinib) en febrero de 2017 con respuesta parcial. La TC de enero de 2018 objetivó el tumor sin progresión radiológica pero con ACTH elevada de 676pg/ml, cortisol sérico de 25,6μg/dl y CLU de 766,7μg/día, en tratamiento con lenvatinib y metopirona.
Caso 2Mujer de 50 años diagnosticada en 2014 de SC por secreción ectópica de ACTH por TNEp bien diferenciado (grado2 con Ki67 del 15%) y metástasis hepáticas irresecables. Presentaba inestabilidad postural, astenia y debilidad muscular proximal. A la exploración física distensión abdominal, hiperpigmentación, hirsutismo y acné. Analíticamente destacaban una concentración plasmática de ACTH de 1.085pg/ml, cortisol sérico de 30μg/dl, CLU de 3.900μg/24h, sin frenar en test de supresión débil y fuerte con dexametasona. La TC mostró un tumor sólido en istmo-cabeza de páncreas con diseminación hepática. La gammagrafía para receptores de somatostatina fue positiva en cabeza de páncreas. Se realizó QE de las metástasis, se colocó endoprótesis en vena porta y para el control del SC se inició ketoconazol y espironolactona. Sin embargo, persistía una concentración plasmática de ACTH de 758,34pg/ml y CLU de 664μg/día, indicándose suprarrenalectomía bilateral (junio de 2015). Ante ello, en enero de 2016 se inició quimioterapia (estreptozocina y 5-fluorouracilo), finalizándose en abril por progresión hepática tras 3 ciclos e iniciándose en mayo sunitinib (37,5g/día).
La TC de enero de 2018 no mostró signos de progresión radiológica, pero sí persistencia de ACTH elevada (4.700pg/ml) y CLU de 132μg/día, en tratamiento sustitutivo con hidroaltesona y fludrocortisona.
DiscusiónEl objetivo del tratamiento del TNEp productor de ACTH irresecable es el control bioquímico del SC y la prevención de la progresión de la enfermedad5. La cromogranina sérica es el marcador general bioquímico para el seguimiento de la progresión tumoral, pero cuando es funcionante el marcador también será la hormona en exceso. Así, en los raros casos de TNEp productores de ACTH como los presentados, se observa una relación paralela entre los niveles de ACTH y de cromogranina según la funcionalidad del tumor (fig. 1). Por otro lado, el mal control del hipercortisolismo es un factor de mal pronóstico2,3,6, que es conveniente controlar previo al inicio de cualquier tratamiento. En los tumores que expresan SSTR-2 y SSTR-5 los análogos de la somatostatina son una estrategia terapéutica para estabilizar el crecimiento y disminuir la secreción hormonal7. Otra opción terapéutica para la hipercortisolemia son la metirapona o el ketoconazol, así como la adrenalectomía bilateral, que ha demostrado mejorar la supervivencia en los primeros dos años2, como en nuestro segundo caso. Para la progresión de la enfermedad en el cáncer avanzado irresecable, everolimus y sunitinib6,7, e incluso lenvatinib, son nuevas opciones esperanzadoras, aunque existe poca experiencia a largo plazo. Everolimus fue empleado en el primer caso sin éxito y sunitinib en el segundo con estabilización de la enfermedad. El tratamiento quimioterápico tradicional con estreptozocina y 5-fluorouracilo es una opción, empleada sin éxito en el segundo caso. La terapia con radionúclidos puede ser eficaz cuando el SRS o 68Ga-DOTA-peptide-PET/CT son positivos2,3.

El cambio de funcionalidad debido a la pluripotencialidad dificulta el manejo. En el primer caso inicialmente no funcionante, desarrolla hiperfunción de ACTH a los tres años, siendo de ayuda la repetición del examen histológico de los depósitos metastásicos, para definir el comportamiento biológico del tumor5.
Podemos concluir que para los TNEp secretores de ACTH faltan decisiones de tratamiento basadas en la evidencia. En la trayectoria de los casos descritos se realizan cambios de tratamiento sin una respuesta eficaz completa. Se requieren ensayos clínicos aleatorizados para establecer las mejores opciones terapéuticas, ya que actualmente la única fuente de datos científicos son las series breves de casos como los expuestos3,4.
Conflicto de interesesLos autores declaran que no tienen ningún tipo de conflicto de intereses.