




El nacimiento de una criatura interesexual provoca una estrategia de manejo de largo plazo que involucra a una miríada de profesionales para trabajar con la familia. Se ha progresado en el diagnóstico, las técnicas quirúrgicas, la comprensión de los problemas psicosociales y el reconocimiento y aceptación del apoyo al paciente. La Sociedad Pediátrica Endocrinológica de Europa consideró oportuno revisar el manejo de los trastornos intersex desde una perspectiva amplia, reseñar información sobre los resultados a largo plazo y formular propuestas para estudios futuros. La metodología incluyó el establecimiento de grupos de trabajo formados a partir de 50 expertos internacionales en el campo. Los grupos prepararon respuestas escritas previas a un cuestionario definido a partir de una revisión de la bibliografía basada en evidencias. En una reunión subsecuente de los participantes, se acordó un documento de consenso. El presente artículo constituye su forma final.
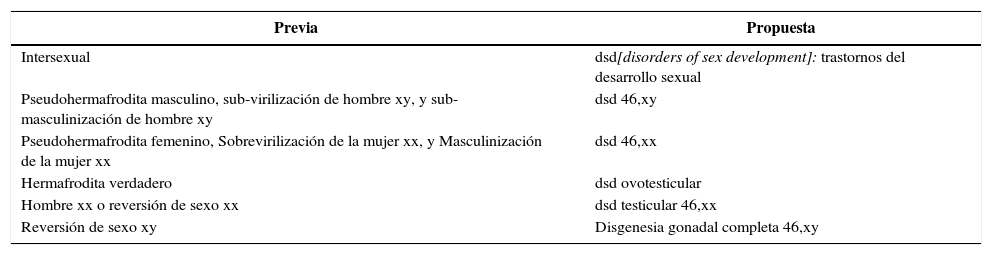
Nomenclatura y definicionesLos avances en la identificación de las causas genéticas moleculares de anormalidades en el sexo, con una conciencia creciente de los problemas éticos y la preocupación por el apoyo al paciente, requieren un nuevo examen de la nomenclatura (Frader, et al. 2004). Términos tales como intersexo, pseudohermafroditismo, hermafroditismo, reversión sexual, así como las etiquetas diagnósticas basadas en el género, son particularmente controvertidos. Estos términos se perciben como potencialmente peyorativos por los pacientes (Conn, Gillam y Conway 2005), y pueden producir confusión tanto en el personal médico como en los progenitores. Proponemos el término “trastornos del desarrollo sexual” (dsd, por sus siglas en inglés)2 y lo definimos como las condiciones congénitas en donde el desarrollo del sexo cromosómico, gonadal o anatómico es atípico. Los cambios propuestos en la terminología se resumen en el cuadro 1. Se necesita un léxico moderno para integrar el progreso en los aspectos genético-moleculares del desarrollo sexual. Como los datos sobre resultados en individuos con dsd son limitados, es esencial la precisión cuando se aplican definiciones y etiquetas diagnósticas (Dreger, Chase, Sousa, Grupposo y Frader 2005; Brown y Warne 2005). También es apropiado usar una terminología sensible a las preocupaciones de los pacientes. La nomenclatura ideal deberá ser tan flexible como para incorporar nueva información que ya sea suficientemente sólida como para mantener un marco consistente. Los términos pueden ser descriptivos y reflejar la etiología genética cuando esté disponible y acomodarse al espectro de la variación fenotípica. Médicos y científicos deben valorar el uso de la nomenclatura, y esta debe ser comprensible para los pacientes y sus familias. En el cuadro 2 se muestra un ejemplo de cómo puede ser aplicada la nomenclatura propuesta en una clasificación de dsd.
Nomenclatura propuesta revisada.
| Previa | Propuesta |
|---|---|
| Intersexual | dsd[disorders of sex development]: trastornos del desarrollo sexual |
| Pseudohermafrodita masculino, sub-virilización de hombre xy, y sub-masculinización de hombre xy | dsd 46,xy |
| Pseudohermafrodita femenino, Sobrevirilización de la mujer xx, y Masculinización de la mujer xx | dsd 46,xx |
| Hermafrodita verdadero | dsd ovotesticular |
| Hombre xx o reversión de sexo xx | dsd testicular 46,xx |
| Reversión de sexo xy | Disgenesia gonadal completa 46,xy |
Ejemplo de una clasificación dsd.
| dsd sexo cromosómico | dsd 46, XY | dsd 46, XX |
|---|---|---|
| 45,x (Síndrome de Turner y sus variantes) | Trastornos del desarrollo gonadal (testicular): 1) disgenesia gonadal completa (síndrome de Swyer); 2) disgenesia gonadal parcial; 3) regresión gonadal; 4) dsd ovotesticular | Trastornos del desarrollo gonadal (ovárico): 1) dsd ovotesticular; 2) dsd testicular (p.ej. sry+, duplicación sox9); 3) disgenesia gonadal |
| 47,xxy (Síndrome de Klinefelter y sus variantes) | Trastornos en la síntesis o acción de los andrógenos: 1) defecto en la biosíntesis de los andrógenos (p.ej. deficiencia de 17-hidroxiesteroide deshidrogenasa; deficiencia de 5ard2, mutaciones star); 2) defecto en la acción de los andrógenos (p. ej. cais, pais); 3) defectos en el receptor de hormona luteinizante (p. ej. hipoplasia de las células de Leydig, aplasia); 4) trastornos de la hormona anti-mulleriana y del receptor de la hormona anti-mulleriana (síndrome del ducto mulleriano persistente) | Exceso de andrógenos: 1) fetal (p.ej. deficiencia de 21 hidroxilasa); 2) feto-placentario (deficiencia de aromatasa, por [oxidoreductasa p450]); 3) materna (luteoma, exógeno, etc.) |
| 45,x/46,xy (Disgenesia gonadal mixta, dsd ovotesticular) | Otros (p.ej. extrofia claocal, atresia vaginal, murcs [anormalidades mullerianas, renales, y de somites cérvico torácicas], otros síndromes) | |
| 46,xx/46,xy (dsd quimérico, ovotesticular) |
[Nota al cuadro: Aunque la consideración del cariotipo es útil para la clasificación, se debe evitar toda referencia innecesaria al cariotipo; idealmente, un sistema basado en términos descriptivos (p.ej. síndrome de insensibilidad a los andrógenos) debe ser usado cuando sea posible. star indica proteína regulatoria esteroideogénica aguda.]
El desarrollo psicosexual se conceptualiza de forma tradicional con tres componentes: la identidad de género se refiere a la autorepresentación de la persona como varón o hembra (con la salvedad de que algunos individuos no pueden ser identificados exclusivamente como uno u otro); el rol de género (conductas típicas de un sexo) describe las características psicológicas que son sexualmente dimórficas dentro de la población general, tales como las preferencias por juguetes y la agresión física; y la orientación sexual se refiere a la dirección (o direcciones) del interés erótico (heterosexual, bisexual, homosexual) e incluye conducta, fantasías y atracciones. El desarrollo psicosexual se ve influido por múltiples factores, tales como la exposición a los andrógenos, los genes de los cromosomas sexuales y la estructura del cerebro, así como las circunstancias sociales y la dinámica familiar. La insatisfacción con el género denota infelicidad con el sexo asignado. Las causas de insatisfacción con el género, incluso entre individuos sin dsd, son escasamente entendidas.
La insatisfacción con el género ocurre más frecuentemente en individuos con dsd que en la población general, pero es difícil predecirla a partir del cariotipo, de la exposición prenatal a los andrógenos, del grado de virilización genital o del género asignado (Cohen-Kettenis 2005; MeyerBahlburg 2005; Zucker 1999). La exposición prenatal a los andrógenos está claramente asociada con otros aspectos del desarrollo psicosexual (Cohen-Bendahan, van de Beek y Berenbaum 2005; Meyer-Bahlburg 2001). Hay efectos relacionados con la dosis en la conducta de juego infantil de niñas con hiperplasia adrenal congénita (cah, por sus siglas en inglés), de modo que aquellas con mutaciones más severas y una virilización genital marcada juegan más que las otras con juguetes masculinos (Nordenström, Servin, Bohlin, Larsson y Wedell 2002). La exposición prenatal a los andrógenos también está asociada con otras características psicológicas, tales como el interés maternal y la orientación sexual. Es importante enfatizar la separa- bilidad de la conducta sexual típica, la orientación sexual y la identidad de género, de modo que la orientación homosexual (relacionada con el sexo de crianza) o un fuerte interés en el otro sexo en individuos con dsd no es un indicador de asignación incorrecta de género. Entender las variaciones en el desarrollo psicosexual en individuos con dsd requiere referirse a estudios en especies no humanas que muestran efectos marcados pero complejos de los andrógenos en la diferenciación sexual del cerebro y la conducta. Los resultados pueden ser influidos por el momento, la dosis y el tipo de exposición al andrógeno, la disponibilidad de un receptor y modificaciones debidas al ambiente social (Goy, Bercovitch y McBrair 1988; Wallen 2005; Moore 1992; Wallen 1996).
Datos provenientes de la investigación en roedores sugieren que los genes del sexo cromosómico pueden también influir directamente en la estructura del cerebro y en la conducta (De Vries, et al. 2002; Skuse et al. 1997). Sin embargo, en ningún estudio sobre individuos con el síndrome de insensibilidad completa a los andrógenos (casi, por sus siglas en inglés) se indica un rol conductual para los genes cromosómicos Y, aunque los datos son limitados (Hines, Ahmed y Hughes 2003), se han identificado diferencias sexuales en las estructuras del cerebro en diferentes especies, y algunas coinciden con el inicio de la pubertad, lo cual podría sugerir respuesta hormonal (Arnold, Rissman y De Vries 2003; Luders et al. 2004; Paus 2005). El sistema límbico y el hipotálamo, cada uno de los cuales tiene un papel en la reproducción, muestran diferencias de sexo en núcleos específicos, pero no está claro cuándo emergen estas diferencias. La interpretación de las diferencias de sexo se complica por el efecto de la muerte celular y la desaferentación neuronal de la maduración normal y por los efectos de la experiencia en el cerebro. La estructura del cerebro en el momento actual no es útil para la asignación del género.
Investigación y manejo de dsdConceptos generalesUn manejo clínico óptimo de individuos con dsd (cmdsd 2006) debería incluir lo siguiente: 1) la asignación de género a recién nacidos debe ser evitada antes de la evaluación de los expertos; 2) la evaluación y el manejo a largo plazo deben ser llevados a cabo en un centro por un equipo multidisciplinario con experiencia; 3) todos los individuos deben recibir una asignación de género; 4) la comunicación abierta con los pacientes y sus familias es esencial, y se debe estimular su participación en la toma de decisiones; y 5) las preocupaciones del paciente y su familia deben ser respetadas y abordadas en estricta confidencialidad.
El primer contacto con los progenitores de una criatura con un dsd es especialmente importante, porque las primeras impresiones de estos encuentros a menudo persisten. Un punto clave a subrayar es que la criatura con un dsd tiene el potencial de convertirse en un integrante de la sociedad bien adaptado y funcional. Aunque la privacidad necesita ser respetada, un dsd no es vergonzoso. Debe explicarse a los progenitores que el curso de acción óptimo puede no ser claro al inicio, pero el equipo médico trabajará con la familia para llegar al mejor conjunto de decisiones posibles en esas circunstancias. El equipo de salud debe discutir con los progenitores qué información compartir en las etapas tempranas con los miembros de la familia y con las amistades. Los progenitores deben estar informados sobre el desarrollo sexual, y la información de internet puede ayudar, siempre y cuando el contenido y el foco de la información estén balanceados y sean sensatos.
Debe darse tiempo y oportunidad amplios para la discusión continua con revisión de la información previamente provista (Frader, et al. 2004).
El equipo multidisciplinarioEl cuidado óptimo para criaturas con dsd requiere de un equipo multidisciplinario experimentado que generalmente se encuentra en centros médicos de tercer nivel. Idealmente, el equipo incluye subespecialistas pediátricos en endocrinología, cirugía y/o urología, psicología/psiquiatría, ginecología, genética, neonatología y, si hay disponibilidad, trabajo social, enfermería y ética médica (Lee 2004). La composición nuclear variará de acuerdo con el tipo de dsd, los recursos locales, el contexto de desarrollo y el lugar. La comunicación constante con el médico familiar de primer nivel de atención es esencial (aap 2005).
El equipo tiene la responsabilidad de educar a otro grupo de personal de salud en el manejo inicial apropiado de recién nacidos afectados y sus familias. Para pacientes nuevos con dsd, el equipo debe desarrollar un plan de manejo clínico relacionado con el diagnóstico, la asignación de género y las opciones de tratamiento, antes de hacer recomendación alguna. Idealmente, se llevan a cabo discusiones con la familia dirigidas por un profesional con habilidades de comunicación apropiadas (Cashman, Reidy, Cody y Lemay 2004). El cuidado transicional debe ser organizado con el equipo multidisciplinario y llevado a cabo en un ambiente que incluya especialistas con experiencia tanto en la práctica pediátrica como en la práctica adulta. Los grupos de apoyo pueden tener un papel importante en el cuidado de pacientes con dsd y sus familias (Warne 2003) (véase el apéndice 1).
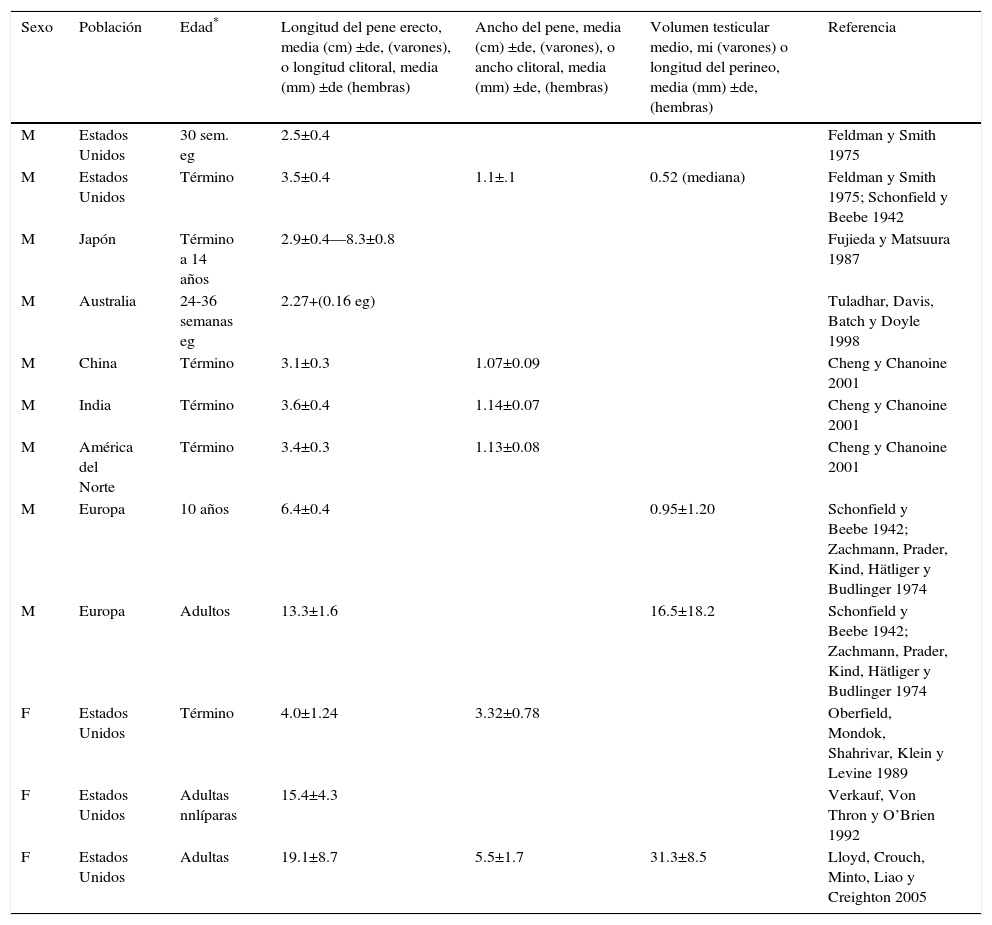
Evaluación clínicaDebe registrarse un expediente con la historia familiar y prenatal, un examen físico general con atención a cualquier característica dimórfica asociada y una evaluación de la anatomía genital que se compare con las normas publicadas (cuadro 3). Los criterios que sugieren dsd incluyen: 1) ambigüedad genital manifiesta (p.ej. extrofia cloacal), 2) genitales aparentemente femeninos con un clítoris alargado, fusión labial posterior o una masa inguinal/ labial, 3) genitales aparentemente masculinos con testículos bilaterales no descendidos, micropene, hipospadias perineal aislado o hipospadias leve con testículos no descendidos, 4) historia familiar de dsd, como la que se da en casi; y 5) discordancia entre la apariencia genital y un cariotipo prenatal. La mayor parte de las causas de dsd se reconocen en el periodo neonatal; presentaciones tardías en criaturas mayores y adultos jóvenes incluyen: 1) ambigüedad genital previamente no reconocida, 2) hernia inguinal en una hembra, 3) pubertad demorada o incompleta, 4) virilización en una hembra, 5) amenorrea primaria, 6) desarrollo de mamas en un varón y 7) hematuria fuerte y ocasionalmente cíclica en un varón.
Mediciones antropométricas de los genitales externos
| Sexo | Población | Edad* | Longitud del pene erecto, media (cm) ±de, (varones), o longitud clitoral, media (mm) ±de (hembras) | Ancho del pene, media (cm) ±de, (varones), o ancho clitoral, media (mm) ±de, (hembras) | Volumen testicular medio, mi (varones) o longitud del perineo, media (mm) ±de, (hembras) | Referencia |
|---|---|---|---|---|---|---|
| M | Estados Unidos | 30 sem. eg | 2.5±0.4 | Feldman y Smith 1975 | ||
| M | Estados Unidos | Término | 3.5±0.4 | 1.1±.1 | 0.52 (mediana) | Feldman y Smith 1975; Schonfield y Beebe 1942 |
| M | Japón | Término a 14 años | 2.9±0.4—8.3±0.8 | Fujieda y Matsuura 1987 | ||
| M | Australia | 24-36 semanas eg | 2.27+(0.16 eg) | Tuladhar, Davis, Batch y Doyle 1998 | ||
| M | China | Término | 3.1±0.3 | 1.07±0.09 | Cheng y Chanoine 2001 | |
| M | India | Término | 3.6±0.4 | 1.14±0.07 | Cheng y Chanoine 2001 | |
| M | América del Norte | Término | 3.4±0.3 | 1.13±0.08 | Cheng y Chanoine 2001 | |
| M | Europa | 10 años | 6.4±0.4 | 0.95±1.20 | Schonfield y Beebe 1942; Zachmann, Prader, Kind, Hätliger y Budlinger 1974 | |
| M | Europa | Adultos | 13.3±1.6 | 16.5±18.2 | Schonfield y Beebe 1942; Zachmann, Prader, Kind, Hätliger y Budlinger 1974 | |
| F | Estados Unidos | Término | 4.0±1.24 | 3.32±0.78 | Oberfield, Mondok, Shahrivar, Klein y Levine 1989 | |
| F | Estados Unidos | Adultas nnlíparas | 15.4±4.3 | Verkauf, Von Thron y O’Brien 1992 | ||
| F | Estados Unidos | Adultas | 19.1±8.7 | 5.5±1.7 | 31.3±8.5 | Lloyd, Crouch, Minto, Liao y Creighton 2005 |
Se han dado progresos considerables en la comprensión de la base genética del desarrollo sexual humano (Grumbach, Hughes y Conte 2003); sin embargo, se identifica un diagnóstico molecular específico en sólo ~20% de los casos de dsd. La mayoría de los infantes virilizados 46,xx tendrán cah. En contraste, sólo 50% de las criaturas 46,xy con dsd recibirán un diagnóstico definitivo (Ahmed, et al. 2000; Morel, et al. 2002). Existen algoritmos diagnósticos, pero, dentro del espectro de diagnósticos y hallazgos, ningún protocolo de evaluación por sí mismo puede ser recomendado en todas las circunstancias. Algunas pruebas, como las imágenes de ultrasonido, son dependientes del operador. La medición de hormonas necesita ser interpretada en relación a las características específicas de la técnica utilizada y con valores normales para la edad gestacional y cronológica. En algunos casos, se necesita hacer mediciones seriadas.
Las pruebas de primera línea en recién nacid*s3 incluyen la de cariotipo con detección de sonda x- e y- específica (incluso cuando el cariotipo prenatalestá disponible), imagen (ultrasonido abdominopélvico), medición de 17-hi- droxiprogesterona, testosterona, gonadotropina, hormona antimülleriana, electrolitos séricos y análisis de orina. Los resultados de estas investigaciones están listos generalmente en 48 horas y serán suficientes para un diagnóstico de trabajo. Se dispone de algoritmos de toma de decisiones para guiar la investigación adicional (Ogilvy-Stuart y Brain 2004). Estas evaluaciones incluyen pruebas de gonadotropina corionica humana y prueba de estimulación adrenocorticotropica para evaluar la biosíntesis testicular y adrenal de esteroides, análisis de esteroides urinarios con espectroscopía de masas por cromatografía de gas, estudios de imagenología y biopsias de material gonadal. Algunos análisis genéticos se llevan a cabo en laboratorios clínicos. No obstante, el diagnóstico molecular corriente está limitado por el costo, la accesibilidad y el control de calidad (Quillin, Jackson-Cook y Bodurtha 2003). Los laboratorios de investigación proveen pruebas genéticas que incluyen análisis funcional, pero pueden enfrentarse a restricciones para la comunicación de resultados (Pagon, et al. 2002).
Asignación de género en recién nacid*sLa incertidumbre inicial sobre el género es perturbadora y estresante para las familias. Hace falta hacer una evaluación meticulosa y tomar una decisión expedita. Los factores que influyen en la asignación de género incluyen el diagnóstico, la apariencia de los genitales, las opciones quirúrgicas, la necesidad de terapia de reemplazo permanente, el potencial para la fertilidad, los puntos de vista de la familia y, a veces, circunstancias relacionadas con las prácticas culturales. Más de 90% de l*s pacientes con cah 46,xx (Dessens, Slijper y Drop 2005) y tod*s l*s pacientes con cais 46,xy que fueron asignad*s como hembras en la infancia (Mazur 2005) se identifican como hembras. La evidencia apoya la recomendación actual de criar a l*s infantes 46,xx marcadamente virilizad*s con cah como hembras (Clayton, et al. 2002). Aproximadamente 60% de l*s pacientes con deficiencia de 5-α-reductasa (5αrd2) que fueron asignad*s como hembras en la infancia y se virilizaron en la pubertad (y tod*s l*s que fueron asignad*s como varones) viven como varones (Cohen-Kettenis 2005). En las deficiencias de 5αrd2 y posiblemente de 17β-hidroxiesteroide deshidrogenasa que se diagnostican en la infancia, la combinación de una identidad de género masculina en la mayoría de edad y el potencial para la fertilidad (documentado respecto de la deficiencia de 5αrd2, pero desconocido en la deficiencia de 17β-hidroxiesteroide deshidrogenasa) deben ser discutidos cuando se aporta evidencia para la asignación de género (Cohen-Kettenis, 2005; Nicolino, Bendelac, Jay, Forest y David 2004; Mendonca, et al. 2003). Entre pacientes con síndrome de insensibilidad parcial a los andrógenos (pais, por sus siglas en inglés), defectos en la biosíntesis de los andrógenos y disgenesia gonadal incompleta, hay insatisfacción con el sexo de crianza en ~25% de l*s individuos criad*s ya sea como varones o como hembras (Migeon, et al. 2002). Datos disponibles apoyan la crianza como varones en todos los pacientes con micropene, tomando en cuenta una satisfacción igual con el género asignado en aquell*s criados varones o hembras, sin necesidad de cirugía, y el potencial para la fertilidad en pacientes criad*s como varones (Mazur 2005). Aquellas personas que toman la decisión para el sexo de crianza de quienes tienen DSD ovotesticular deben considerar el potencial para la fertilidad sobre la base de la diferenciación gonadal y el desarrollo genital, y deben asumir que los genitales sean, o puedan volverse, consistentes con el sexo elegido. En el caso de disgenesia gonadal mixta (mgd, por sus siglas en inglés), los factores a considerar incluyen la exposición prenatal a andrógenos, la función testicular durante y después de la pubertad, el desarrollo fálico y la localización gonadal. L*s pacientes con extrofia cloacal criad*s como hembras muestran variabilidad en el resultado de la identidad de género, pero, aparentemente, >65% viven como hembras (Meyer-Bahlburg 2005).
Manejo quirúrgicoEl cirujano tiene la responsabilidad de explicar la secuencia quirúrgica y sus consecuencias subsecuentes desde la infancia hasta la adultez. Sólo cirujanos con pericia en el cuidado de criaturas y con entrenamiento específico en cirugía de dsd deberían llevar a cabo estos procedimientos. Los progenitores ahora parecen menos inclinados a elegir la cirugía para clitoromegalia poco severa (Lee y Witchel 2002). La cirugía sólo debe ser considerada en casos de virilización severa (Prader iii-v) y llevarse a cabo en conjunción, cuando es apropiado, con la reparación del seno urogenital común. Dado que la función orgásmica y la sensación eréctil pueden ser perturbadas por la cirugía clitoridea, el procedimiento quirúrgico debe basarse anatómicamente para preservar la función eréctil y la innervación del clítoris. Se privilegia el resultado funcional sobre la apariencia estrictamente cosmética. Existe un sentimiento extendido de que la cirugía que se realiza por razones cosméticas en el primer año de vida alivia la angustia parental y mejora los lazos entre la criatura y sus progenitores (Rink y Adams 1998; Farkas, Chertin y Hadas-Halpren 2001; Baskin 2004; Crouch, Minto, Laio, Woodhouse y Creighton 2004); sin embargo, no hay evidencia sistemática que sustente esta creencia.
En la actualidad, hay evidencia inadecuada en relación con el establecimiento de una anatomía funcional para abandonar la práctica de la separación temprana de la vagina y la uretra (Meyer-Bahlburg, et al. 2004). La explicación para la reconstrucción temprana se basa en guías para cirugía genital de la American Academy of Pediatrics (AAP 1996), los efectos benéficos de los estrógenos en los tejidos en la infancia temprana y la evitación de complicaciones potenciales provenientes de la conexión entre el tracto urinario y el peritoneo, vía las trompas de Falopio. Se anticipa que la reconstrucción en la infancia necesitará ser refinada en la pubertad (Eroglu, et al. 2004; Alizai, Thomas, Lilford, Batchelor y Johnson 1999; Bailez, Gearhart, Migeon y Rock 1992). La dilatación vaginal no deberá ser realizada antes de la pubertad. El cirujano debe familiarizarse con una cantidad de técnicas operativas para reconstruir el espectro de los desórdenes del seno urogenital. Una vagina ausente o inadecuada (con raras excepciones) requiere vaginoplastia durante la adolescencia, cuando la paciente está motivada en términos psicológicos y participa plenamente en el procedimiento. Ninguna técnica ha sido exitosa a nivel global; la autodilatación, la sustitución con piel y la vaginoplastia con intestino tienen cada una ventajas y desventajas específicas.
En el caso de un dsd asociado con hipospadias (Mouriquand y Mure 2004), se aplican técnicas estándar para reparación quirúrgica, tales como la corrección de cuerdas, la reconstrucción uretral y el uso razonable de testos- terona. La magnitud y complejidad de la faloplastia en la edad adulta debe tomarse en cuenta durante el periodo inicial de consulta si una asignación de género exitosa depende de este procedimiento (Bettocchi, Ralph y Pryor 2005). A veces, esto puede afectar el balance de la asignación de género. No se debe dar a l*s pacientes expectativas poco realistas sobre la reconstrucción peneana, incluido el uso de ingeniería de tejidos. No hay evidencia de que se requiera la remoción profiláctica de estructuras discordantes asintomáticas, como un utrículo o remanentes müllerianos, aunque los síntomas en el futuro podrían indicar la remoción quirúrgica. Para el varón que ha tenido una neofaloplastia exitosa en la edad adulta, puede insertarse una prótesis eréctil, pero tiene alta morbilidad.
Los testículos, en pacientes criados como hembras con cais (Grumbach, Hughes y Conte 2003) o con pais, serían removidos para prevenir malignidad en la vida adulta. La disponibilidad de terapia de reemplazo de estrógenos permite la opción de una extracción temprana en el momento del diagnóstico y también atender la hernia asociada, problemas psicológicos por la presencia de testículos y el riesgo de malignidad. La elección parental puede aplazarse hasta la adolescencia, reconociendo que el reporte más temprano de malignidad en cais se ha dado a los 14 años de edad (Hurt, Bodurtha, McCall y Ali 1989). La estría gonadal en un paciente con mgd criado como varón debe removerse laparoscópicamente (o por laparotomía) en la infancia temprana (Grumbach, Hughes y Conte 2003). La gonadecto- mía bilateral se lleva a cabo en la infancia temprana en hembras (estrías gonadales bilaterales) con disgenesia gonadal y material cromosómico Y. En pacientes criadas como hembras con defectos biosintéticos de los andrógenos, la gonadectomía sería llevada a cabo antes de la pubertad. Un testículo escrotal en pacientes con disgenesia gonadal está en riesgo de malignidad. Las recomendaciones corrientes son la biopsia testicular en la pubertad para buscar signos de la lesión premaligna, denominada carcinoma in situ o neoplastia intratubular indiferenciada de células germinales. Si la prueba es positiva, la opción es crear un banco de esperma antes del tratamiento con radioterapia de dosis baja local, la cual es curativa (Rorth, et al. 2000).
El manejo quirúrgico en dsd debe también considerar opciones que facilitarán la posibilidad de fertilidad. En pacientes con un utrículus sintomático, la extracción se lleva a cabo mejor laparoscópicamente para incrementar la posibilidad de preservar la continuidad de conductos deferentes. Pacientes con ovotestes bilaterales son potencialmente fértiles por el tejido ovárico funcional (Grumbach, Hughes y Conte 2003; Nihoul-Fékéte 2004). La separación de los tejidos ováricos y testiculares puede ser técnicamente difícil y debe ser llevada a cabo, si es posible, a una edad temprana.
Reemplazo de esteroides sexualesEl hipogonadismo es común en pacientes con gónadas disgenéticas, defectos en la biosíntesis de esteroides sexuales y resistencia a los andrógenos. El momento de inicio de la pubertad puede variar, pero es una ocasión que aporta oportunidad para discutir la condición y sienta las bases para una adhesión terapéutica a largo plazo. La inducción hormonal en la pubertad estimula la réplica de maduración de pubertad normal al inducir caracteres sexuales secundarios, una racha de crecimiento de pubertad y una acumulación mineral óptima en los huesos, así como apoyo psicosocial para la maduración psicosexual (Warne, Grover y Zajac 2005). Inyecciones intramusculares de esteres de testosterona se usan comúnmente en varones; otra opción es el undecanoato de testosterona oral, y también hay preparaciones transdérmicas (Rogol 2005; Ahmed, Tucker, Mayo, Wallace y Hughes 2004). Pacientes con PAIS pueden requerir dosis suprafisiológicas de testosterona para un resultado óptimo (Weidemann, Peters, Romalo, Spidler y Schweikert 2000). Las hembras con hipogonadismo requieren un suplemento de estrógeno para inducir cambios de pubertad y menstruación. Un progestageno se añade por lo regular después del primer sangrado o al año o dos años de estrógeno continuo. No hay evidencia de que la adición de progesterona cíclica sea benéfica en mujeres sin útero.
Manejo psicosocialEl cuidado psicosocial que aporta el equipo de salud mental especializado en dsd debe ser una parte integral del manejo para promover una adaptación positiva. Esta especialización puede facilitar las decisiones del equipo sobre la asignación o reasignación de género, los momentos para la cirugía y el reemplazo sexo-hormonal. Se cuenta con herramientas para detección psicosocial que identifican a las familias en riesgo de tener una adaptación deficiente a la condición médica de una criatura (Kazak, et al. 2003). Una vez que la criatura está suficientemente desarrollada para una evaluación de identidad de género, tal evaluación debe incluirse en las discusiones sobre la reasignación de género. El desarrollo de la identidad de género inicia antes de los tres años de edad (Martin, Ruble y Szkrybalo 2002), pero la etapa más temprana en que puede ser evaluada confiablemente todavía no es clara. La generalización de que los 18 meses es el límite superior para la imposición de una reasignación de género debe ser tratada con precaución y contemplada de manera conservadora. Una conducta de rol de género atípica es más común en criaturas con dsd que en la población general, pero no debe ser tomada como un indicador para la reasignación de género. En criaturas y adolescentes afectados que reportan disforia de género significativa, se requiere una evaluación psicológica comprehensiva (Zucker 2005) y una oportunidad para explorar sus sentimientos sobre el género con un especialista calificado en un determinado lapso. Si el deseo de cambiar de género persiste, la opinión del/la paciente debe ser apoyada y puede requerir la participación de un especialista calificado en el manejo de cambio de género.
El proceso de apertura respecto de hechos sobre cariotipo, estatus gonadal y prospectos para la fertilidad futura es una acción colaborativa y continua que requiere una aproximación flexible e individualizada. Debe planearse con los progenitores desde el momento del diagnóstico (Carmichael y Ransley 2002). Estudios sobre otros trastornos médicos crónicos y sobre hijos adoptados indican que la apertura se asocia con una mejor adaptación psicosocial (aap 1999). La educación y la consejería médica para las criaturas es un proceso gradual y recurrente de refinamiento creciente que se adecua al cambio cognitivo y al desarrollo psicológico (Money 1994).
La calidad de vida incluye enamorarse, salir, atraer, la capacidad para desarrollar relaciones íntimas, el funcionamiento sexual y la oportunidad de casarse y tener descendencia, no importa cuáles sean los indicadores biológicos del sexo. Los problemas más frecuentes encontrados en pacientes con dsd son la aversión al sexo y la falta de excitabilidad, que a menudo son malinterpretadas como ausencia de libido (Basson, et al. 2003). El equipo de cuidado de salud debe ofrecer a l*s pacientes en la adolescencia la oportunidad de hablar confidencialmente, sin la presencia de sus progenitores, y estimular su participación en grupos de apoyo específicos que amplían su capacidad para discutir sobre sus preocupaciones con comodidad. Algun*s pacientes evitan las relaciones íntimas, y es importante abordar su miedo al rechazo y aconsejarlos en el proceso de construir una relación de pareja. El foco debe ser sobre las relaciones interpersonales y no sólo sobre la función y actividad sexual. Puede requerirse referencia a terapia sexual. Los repetidos exámenes de los genitales, que incluyen fotografía médica, pueden ser experimentados como algo profundamente vergonzoso (Creighton, Alderson, Brown y Minto 2002). La fotografía médica tiene un papel informativo y educativo, pero debe ser llevada a cabo, siempre que sea posible, cuando l*s pacientes están anestesiad*s para algún procedimiento. Algunas intervenciones médicas y experiencias sexuales negativas han producido síntomas del trastorno de estrés postraumático, en cuyo caso l*s pacientes deben ser canalizad*s a profesionales calificados en salud mental (Ursano, et al. 2004).
Resultados en dsdEn términos generales, hay insuficiente información en una amplia gama de evaluaciones en dsd. Lo que sigue está basado en aquellos desórdenes para los cuales hay alguna evidencia básica disponible. Incluyen cah, cais y pais, desórdenes de la biosíntesis de los andrógenos, síndromes de disgenesia gonadal (completa y parcial) y micropene. Los resultados a largo plazo en dsd deben incluir el fenotipo genital externo e interno, salud física (incluso fertilidad, función sexual) y ajuste social y psicosexual, salud mental, calidad de vida y participación social. Hay problemas de salud adicionales en personas con dsd que incluyen las consecuencias de problemas asociados, tales como otras malformaciones, retraso en el desarrollo y deterioro intelectual, crecimiento y desarrollo demorados y efectos indeseados de las hormonas en la libido y la imagen corporal (Kuhnle y Bullinger 1997).
Resultado quirúrgicoAlgunos estudios sugieren resultados satisfactorios de cirugías tempranas (Clayton, et al. 2002; Migeon, et al. 2002;Lee y Witchel 2002; Warne, et al. 2005). No obstante, resultados de clitoriplastías identifican problemas relacionados con disminución en la sensibilidad sexual, pérdida de tejido clitoral y problemas cosméticos (Creighton 2004). Algunas técnicas para la vaginoplastia acarrean potencial de escarificar el introitus, por lo que requieren modificaciones repetidas antes de que la función sexual sea confiable. La cirugía para construir una neovagina acarrea el riesgo de neoplasia (Steiner y Woernie 2002). Los riesgos de la vaginoplastia son diferentes para la alta y la baja confluencia de la uretra y la vagina. Es complicado realizar análisis de resultados de largo plazo por una mezcla de técnicas quirúrgicas y categorías diagnósticas (Schober 2006). Pocas mujeres con cais necesitan cirugía para ampliar la vagina (Wisniewski, et al. 2000).
El resultado en varones poco masculinizados con falo depende del grado de hipospadias y del monto de tejido eréctil. La genitoplastia feminizante, en contraste con la genitoplastia masculinizante, requiere menos cirugía para llegar a un resultado aceptable y deriva en menos dificultades urológicas (Migeon, et al. 2002). Los datos a largo plazo sobre función sexual y calidad de vida para personas asignados como hembras y para personas asignadas como varones muestran gran variabilidad. No hay pruebas clínicas controladas de la eficacia de cirugías tempranas (antes de los 12 meses de edad) o tardías (en la adolescencia o adultez) o de la eficacia de técnicas diferentes.
Riesgo de tumores gonadalesEn la bibliografía especializada, la interpretación del riesgo de tumores gonadales es entorpecida por una terminología poco clara, y por los efectos de demora en la maduración celular normal (Honecker, Stoop, de Krijger, Chris Lau, Bokemeyer y Looijenga 2004; Cools, et al. 2005; Cools, et al. 2006). El mayor riesgo de tumor se encuentra en (codificación de proteína Y específica testicular) disgénesis gonadal positiva y en PAIS con gónadas intrabdominales, mientras que el menor riesgo (<5%) se encuentra en ovotestis (Ramani, Yeung y Habeebu 1993) y casi (Cools, et al. 2005; Hannema, Scott, Rajperts-De Meyts, Skakkebaek, Coleman y Hughes 2006). El cuadro 4 aporta un resumen del riesgo de desarrollo de tumores de acuerdo con el diagnóstico y las recomendaciones para su manejo.
Riesgo de malignidad de células germinales de acuerdo con el diagnóstico.
| Grupo de riesgo | Trastorno | Riesgo de malignidad, % | Acción recomendada | Pacientes (n) | Estudios (n) |
|---|---|---|---|---|---|
| Alto | dg{a} (+y){b} intrabdominal | 15-35 | Gonadectomía{c} | 12 | <350 |
| pais no escrotal | 50 | Gonadectomía{c} | 2 | 24 | |
| Frasier | 60 | Gonadectomía{c} | 1 | 15 | |
| Denys-Drash (+y) | 40 | Gonadectomía{c} | 1 | 5 | |
| Intermedio | Turner (+y) | 12 | Gonadectomía{c} | 11 | 43 |
| 17 β-hidroxiesteroides | 28 | Espera de observación | 2 | 7 | |
| dg (+y){b} escrotal | Desconocido | ¿Biopsia{d} e irradiación? | 0 | 0 | |
| pais gónada escrotal | Desconocido | ¿Biopsia{d} e irradiación? | 0 | 0 | |
| Bajo | cais | 2 | ¿Biopsia{d} y ??? | 2 | 55 |
| dsd ovotesticular | 3 | ¿Remoción de tejido testicular? | 3 | 426 | |
| Turner (-y) | 1 | Ninguno | 11 | 557 | |
| Ninguno (?) | 5αrd2 | 0 | No resuelto | 1 | 3 |
| Hipoplasia de células de Leydig | 0 | No resuelto | 1 | 2 |
Los dsd pueden acarrear un estigma. Factores sociales y culturales, así como los efectos hormonales, parecen influir en el rol de género ante la deficiencia de 5αrd2. El cambio en el rol de género ocurre en diferentes sociedades y en diferentes medidas, lo cual sugiere que los factores sociales también pueden ser modificadores importantes de cambio de rol de género.
En algunas sociedades, la esterilidad femenina impide el matrimonio, lo cual también afecta las perspectivas de empleo y crea dependencia económica. Las perspectivas religiosas y filosóficas pueden influir en cómo responden los progenitores al nacimiento de una criatura con una condición médica. El fatalismo y los sentimientos de culpa relativos a malformaciones congénitas o condiciones genéticas tienen influencia, mientras que la pobreza y la ignorancia afectan negativamente el acceso al cuidado de la salud (Warne y Bhatia 2006).
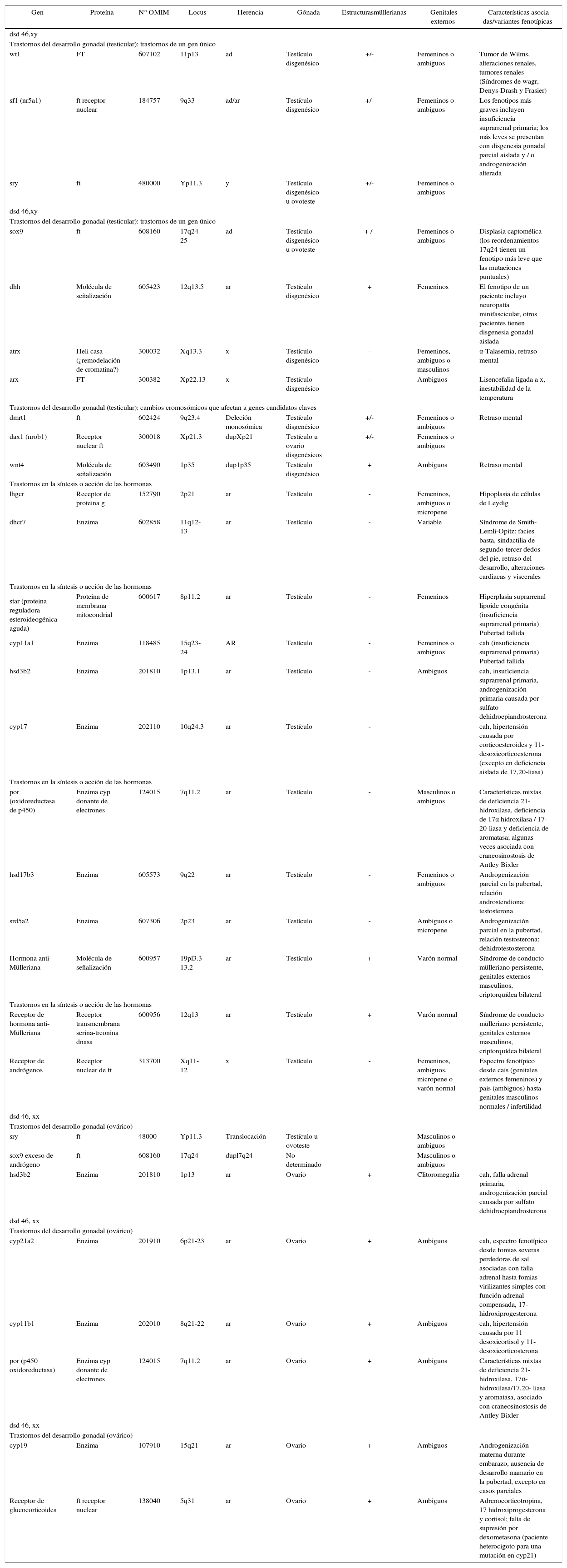
Estudios futurosEstablecer un diagnóstico preciso en dsd es tan importante aquí como en otras condiciones médicas crónicas que tienen consecuencias de por vida. Se ha logrado un progreso considerable con los estudios moleculares, como se ilustra en el cuadro 5, que resume cuáles genes se sabe que tienen que ver con los dsd. El uso de modelos knock-out de tejidos animales específicos, la hibridación genómica comparativa y las pruebas de micro-rna de la cresta urogenital del ratón aportarán beneficios en la identificación de nuevos genes que causan dsd (Small, Shima, Uzumcu, Skinner y Griswold 2005). Es esencial que el impulso de un enfoque colaborativo internacional para esta tarea se sostenga.
Genes conocidos involucrados en dsd.
| Gen | Proteína | N° OMIM | Locus | Herencia | Gónada | Estructurasmüllerianas | Genitales externos | Características asocia das/variantes fenotípicas |
|---|---|---|---|---|---|---|---|---|
| dsd 46,xy | ||||||||
| Trastornos del desarrollo gonadal (testicular): trastornos de un gen único | ||||||||
| wt1 | FT | 607102 | 11p13 | ad | Testículo disgenésico | +/- | Femeninos o ambiguos | Tumor de Wilms, alteraciones renales, tumores renales (Síndromes de wagr, Denys-Drash y Frasier) |
| sf1 (nr5a1) | ft receptor nuclear | 184757 | 9q33 | ad/ar | Testículo disgenésico | +/- | Femeninos o ambiguos | Los fenotipos más graves incluyen insuficiencia suprarrenal primaria; los más leves se presentan con disgenesia gonadal parcial aislada y / o androgenización alterada |
| sry | ft | 480000 | Yp11.3 | y | Testículo disgenésico u ovoteste | +/- | Femeninos o ambiguos | |
| dsd 46,xy | ||||||||
| Trastornos del desarrollo gonadal (testicular): trastornos de un gen único | ||||||||
| sox9 | ft | 608160 | 17q24-25 | ad | Testículo disgenésico u ovoteste | + /- | Femeninos o ambiguos | Displasia captomélica (los reordenamientos 17q24 tienen un fenotipo más leve que las mutaciones puntuales) |
| dhh | Molécula de señalización | 605423 | 12q13.5 | ar | Testículo disgenésico | + | Femeninos | El fenotipo de un paciente incluyo neuropatía minifascicular, otros pacientes tienen disgenesia gonadal aislada |
| atrx | Heli casa (¿remodelación de cromatina?) | 300032 | Xq13.3 | x | Testículo disgenésico | - | Femeninos, ambiguos o masculinos | α-Talasemia, retraso mental |
| arx | FT | 300382 | Xp22.13 | x | Testículo disgenésico | - | Ambiguos | Lisencefalia ligada a x, inestabilidad de la temperatura |
| Trastornos del desarrollo gonadal (testicular): cambios cromosómicos que afectan a genes candidatos claves | ||||||||
| dmrt1 | ft | 602424 | 9q23.4 | Deleción monosómica | Testículo disgenésico | +/- | Femeninos o ambiguos | Retraso mental |
| dax1 (nrob1) | Receptor nuclear ft | 300018 | Xp21.3 | dupXp21 | Testículo u ovario disgenésicos | +/- | Femeninos o ambiguos | |
| wnt4 | Molécula de señalización | 603490 | 1p35 | dup1p35 | Testículo disgenésico | + | Ambiguos | Retraso mental |
| Trastornos en la síntesis o acción de las hormonas | ||||||||
| lhgcr | Receptor de proteina g | 152790 | 2p21 | ar | Testículo | - | Femeninos, ambiguos o micropene | Hipoplasia de células de Leydig |
| dhcr7 | Enzima | 602858 | 11q12-13 | ar | Testículo | - | Variable | Síndrome de Smith-Lemli-Opitz: facies basta, sindactilia de segundo-tercer dedos del pie, retraso del desarrollo, alteraciones cardiacas y viscerales |
| Trastornos en la síntesis o acción de las hormonas | ||||||||
| star (proteina reguladora esteroideogénica aguda) | Proteina de membrana mitocondrial | 600617 | 8p11.2 | ar | Testículo | - | Femeninos | Hiperplasia suprarrenal lipoide congénita (insuficiencia suprarrenal primaria) Pubertad fallida |
| cyp11a1 | Enzima | 118485 | 15q23-24 | AR | Testículo | - | Femeninos o ambiguos | cah (insuficiencia suprarrenal primaria) Pubertad fallida |
| hsd3b2 | Enzima | 201810 | 1p13.1 | ar | Testículo | - | Ambiguos | cah, insuficiencia suprarrenal primaria, androgenización primaria causada por sulfato dehidroepiandrosterona |
| cyp17 | Enzima | 202110 | 10q24.3 | ar | Testículo | - | cah, hipertensión causada por corticoesteroides y 11-desoxicorticoesterona (excepto en deficiencia aislada de 17,20-liasa) | |
| Trastornos en la síntesis o acción de las hormonas | ||||||||
| por (oxidoreductasa de p450) | Enzima cyp donante de electrones | 124015 | 7q11.2 | ar | Testículo | - | Masculinos o ambiguos | Características mixtas de deficiencia 21-hidroxilasa, deficiencia de 17α hidroxilasa / 17-20-liasa y deficiencia de aromatasa; algunas veces asociada con craneosinostosis de Antley Bixler |
| hsd17b3 | Enzima | 605573 | 9q22 | ar | Testículo | - | Femeninos o ambiguos | Androgenización parcial en la pubertad, relación androstendiona: testosterona |
| srd5a2 | Enzima | 607306 | 2p23 | ar | Testículo | - | Ambiguos o micropene | Androgenización parcial en la pubertad, relación testosterona: dehidrotestosterona |
| Hormona anti- Mülleriana | Molécula de señalización | 600957 | 19pl3.3-13.2 | ar | Testículo | + | Varón normal | Síndrome de conducto mülleriano persistente, genitales externos masculinos, criptorquídea bilateral |
| Trastornos en la síntesis o acción de las hormonas | ||||||||
| Receptor de hormona anti- Mülleriana | Receptor transmembrana serina-treonina dnasa | 600956 | 12q13 | ar | Testículo | + | Varón normal | Síndrome de conducto mülleriano persistente, genitales externos masculinos, criptorquídea bilateral |
| Receptor de andrógenos | Receptor nuclear de ft | 313700 | Xq11-12 | x | Testículo | - | Femeninos, ambiguos, micropene o varón normal | Espectro fenotípico desde cais (genitales externos femeninos) y pais (ambiguos) hasta genitales masculinos normales / infertilidad |
| dsd 46, xx | ||||||||
| Trastornos del desarrollo gonadal (ovárico) | ||||||||
| sry | ft | 48000 | Yp11.3 | Translocación | Testículo u ovoteste | - | Masculinos o ambiguos | |
| sox9 exceso de andrógeno | ft | 608160 | 17q24 | dupl7q24 | No determinado | Masculinos o ambiguos | ||
| hsd3b2 | Enzima | 201810 | 1p13 | ar | Ovario | + | Clitoromegalia | cah, falla adrenal primaria, androgenización parcial causada por sulfato dehidroepiandrosterona |
| dsd 46, xx | ||||||||
| Trastornos del desarrollo gonadal (ovárico) | ||||||||
| cyp21a2 | Enzima | 201910 | 6p21-23 | ar | Ovario | + | Ambiguos | cah, espectro fenotípico desde fomias severas perdedoras de sal asociadas con falla adrenal hasta fomias virilizantes simples con función adrenal compensada, 17-hidroxiprogesterona |
| cyp11b1 | Enzima | 202010 | 8q21-22 | ar | Ovario | + | Ambiguos | cah, hipertensión causada por 11 desoxicortisol y 11-desoxicorticosterona |
| por (p450 oxidoreductasa) | Enzima cyp donante de electrones | 124015 | 7q11.2 | ar | Ovario | + | Ambiguos | Características mixtas de deficiencia 21-hidroxilasa, 17α-hidroxilasa/17,20- liasa y aromatasa, asociado con craneosinostosis de Antley Bixler |
| dsd 46, xx | ||||||||
| Trastornos del desarrollo gonadal (ovárico) | ||||||||
| cyp19 | Enzima | 107910 | 15q21 | ar | Ovario | + | Ambiguos | Androgenización materna durante embarazo, ausencia de desarrollo mamario en la pubertad, excepto en casos parciales |
| Receptor de glucocorticoides | ft receptor nuclear | 138040 | 5q31 | ar | Ovario | + | Ambiguos | Adrenocorticotropina, 17 hidroxiprogesterona y cortisol; falta de supresión por dexometasona (paciente heterocigoto para una mutación en cyp21) |
omim: Online Mendelian Inheritance in Man; ft: factor de transcripción; ad: autosómico dominante (frecuente mutación de novo); ar: indica autosómico recesivo; y: cromosoma y; x: cromosoma x. Las reordenaciones cromosómicas incluyen probablemente genes clave. Modiicado de J.C. Achermann, G. Ozisik, J.J. Meeks y J.L, Jameson, 2002, “Genetic causes of human reproductive disease”, Journal of Clinical Endocrinology and Metabolism 87, pp. 2447-2454.
Queda mucho por clarificar sobre los determinantes de la identidad de género en dsd. Los estudios futuros requieren muestreo representativo para conceptualizar y medir con cuidado la identidad de género, reconociendo que hay determinantes múltiples que considerar, y que puede cambiar en la madurez. En términos de manejo psicológico, se necesitan estudios para evaluar la efectividad del manejo de la información en lo que respecta a los tiempos y el contenido. El patrón de la práctica quirúrgica en dsd está cambiando respecto a los tiempos y las técnicas usadas. Es esencial evaluar los efectos de la cirugía temprana versus la tardía de manera holística, reconociendo las dificultades que plantea una práctica clínica en constante evolución.
El consenso ha identificado claramente una enrome carencia de información sobre los resultados de largo plazo. Los estudios futuros deben usar instrumentos apropiados para evaluar los resultados de manera estandarizada (Martin, Ruble y Szkrybalo 2002; Zucker 2005) y tomar en cuenta los lineamientos relevantes de todas las condiciones crónicas.4Estos estudios serían preferentemente de índole prospectiva y estarían diseñados para evitar una selección sesgada. Una cantidad de países ya tiene registros de casos de dsd, pero sería más benéfico compartir tales recursos para permitir estudios prospectivos multicentrados que se llevaran a cabo a partir de un mayor número de casos claramente definidos. Un aliado de esto sería un programa educativo que garantizara que el personal que atiende a las familias donde hay una criatura con dsd estuviera adecuadamente entrenado multiprofesionalmente para aliviar sus responsabilidades.
Apéndice 1: el papel de los grupos de apoyoEl valor del apoyo de pares y parientes en muchas condiciones médicas está ampliamente reconocido, y los dsd, que son condiciones de por vida que afectan el desarrollo en muchas etapas, no son la excepción. Aquellas personas afectadas por dsd y sus parientes valoran lo siguiente:
- •
El apoyo de pares termina con el aislamiento y el estigma, aportando un contexto en que las condiciones se ponen en perspectiva y los problemas íntimos preocupantes pueden ser discutidos en un ambiente de seguridad con alguien que “ya ha estado ahí”.
- •
Las criaturas que establecen relaciones con pares y con adultos afectados desde temprano en la vida se benefician de un sentimiento de normalidad, y cuentan con apoyo desde antes de la adolescencia. Los adolescentes a menudo se resisten a los intentos de introducirlos en instancias de apoyo de pares.
- •
Los grupos de apoyo pueden ayudar a los familiares a encontrar cuidados de la mejor calidad.
Aunque la práctica clínica puede enfocarse en el género y en la apariencia genital como resultados clave, el estigma y las experiencias asociadas con padecer una dsd (tanto dentro como fuera del ambiente médico) son problemas más importantes para la mayoría de la gente afectada.
Los grupos de apoyo complementan el trabajo de los equipos de atención a la salud y, junto con estos, pueden ayudar a mejorar los servicios. Algunas iniciativas de los grupos de apoyo han conducido a mejoras en el manejo de dsd y han dirigido la investigación hacia problemas clínicos relevantes. El diálogo entre los profesionales de atención de la salud y los grupos de apoyo, y su colaboración como socios, deben ser impulsados.
Apéndice 2: problemas legalesLos principios básicos de la ley médica permanecen incluso cuando la investigación y la experiencia clínica evolucionan respecto de la etiología, el diagnóstico y el tratamiento. Este apéndice discute la práctica en tres países de estándares de negligencia médica y consentimiento informado de los pacientes. En Estados Unidos, la profesión médica establece estándares de atención sobre la base de la costumbre médica prevaleciente (Martin 2002). No obstante, un tratamiento también puede ser aquel usado por una minoría respetada de practicantes.
El consentimiento informado en Estados Unidos se fundó sobre el principio del maltrato, dado que es una ofensa violar la integridad corporal de otra persona sin su consentimiento. Hoy en día, la mayoría de los estados de la Unión están preocupados con la revelación negligente de información al paciente. El estándar de revelación adecuada de información puede estar basada en el médico, y debe ser conducida por un médico razonable, o puede estar basada en el paciente, preguntando qué material encontraría un paciente razonable. La revelación basada en el médico debe incluir información sobre riesgos, alternativas, resultados y prognosis, con o sin tratamiento.
Las cortes en Estados Unidos asumen que los progenitores saben qué es lo mejor para su criatura cuando la autoridad parental se aplica al consentimiento para la criatura (juicio sustituto). Las decisiones de los progenitores se omiten en las situaciones en que se aplica un tratamiento para salvar la vida. El consentimiento para el tratamiento de una criatura depende de la comprensión de su índole y consecuencias.
La negligencia médica en el Reino Unido define el tratamiento que está por debajo del estándar esperado de un desempeño razonablemente competente del médico. El estándar de prueba en la corte es si la negligencia es demostrada en un balance de probabilidades. Es obligatorio que el médico demuestre que el tratamiento fue consistente con un conjunto racionalmente defendible de opiniones médicas. Un cambio en la prerrogativa parental al consentimiento se reflejó en una ley de 1989 (Children Act) donde los derechos parentales fueron reemplazados por las responsabilidades parentales. Las cortes del Reino Unido pueden intervenir con órdenes para prevenir una acción específica relacionada con la criatura. La edad no es una barrera para el consentimiento informado, si un menor demuestra suficiente comprensión de los problemas como para tener la capacidad de decidir.
La ley colombiana es notable por un razonable conjunto de lineamien- tos estipulados por la Suprema Corte en casos de dsd (Suprema Corte de Justicia de Colombia 1999). Se formuló un protocolo para la intervención parental y médica. El proceso de consentimiento requiere “consentimiento informado, calificado y persistente” durante un lapso extendido. La autorización se da en etapas para dar tiempo a los progenitores de que se reconcilien con la condición de la criatura. La corte pretende establecer un equilibrio entre la autonomía parental de aquellos que quieren y de aquellos que no quieren cirugía temprana para su criatura hasta que no sea clara la evidencia de daño si se aplaza, mientras la criatura es competente para decidir. Los progenitores no pueden consentir por una criatura mayor de cinco años, porque, para entonces, se considera que la criatura se ha identificado con un género y, por lo tanto, se considera autónoma.
La Lawson Wilkins Pediatric Endocrine Society y la European Society for Paediatric Endocrinology agradece el apoyo irrestricto para la reunión del consenso de Pfizer Endocrine Care, Novo Nordisk, Ferring y Organon.
Las personas participantes en la Conferencia Internacional sobre Intersexo fueron: John Achermann (Londres, Reino Unido), Faisal Ahmed (Glasgow, Reino Unido), Laurence Baskin (San Francisco, California), Sheri Berenbaum (University Park, Pensilvania), Sylvano Bertelloni (Pisa, Italia), John Brock (Nashville, Tennessee), Polly Carmichael (Londres, Reino Unido), Cheryl Chase (Rohnert Park, California), Peggy Cohen-Kettenis (Ámsterdam, Países Bajos), Felix Conte (San Francisco, California), Kenneth Copeland (Oklahoma City, Oklahoma), Patricia Donohoue (Iowa City, Iowa), Chris Driver (Aberdeen, Reino Unido), Stenvert Drop (Rotterdam, Países Bajos), Erica Eugster (Indianapolis, Indiana), Kenji Fujieda (Asahikawa, Japón), Jay Giedd (Bethesda, Maryland), Richard Green (Londres, Reino Unido), Melvin Grumbach (San Francisco, California), Vincent Harley (Victoria, Australia), Melissa Hines (Londres, Reino Unido), Olaf Hiort (Lübeck, Alemania), Christopher Houk (Hershey, Pensilvania), Ieuan Hughes (Cambridge, Reino Unido), Peter Lee (Hershey, Pensilvania), Leendert Looijenga (Rotterdam, Países Bajos), Bernadice Mendoca (Sao Paulo, Brasil), Heino Meyer-Bahlburg (Nueva York, Nueva York), Claude Migeon (Baltimore, Maryland), Yves Morel (Lyon, Francia), Pierre Mouriquand (Lyon, Francia), Anna Nordenström (Estocolmo, Suecia), Phillip Ransley (Londres, Reino Unido), Robert Rapaport (Nueva York, Nueva York), William Reiner (Oklahoma City, Oklahoma), Hertha Richter-Appelt (Hamburg, Alemania), Richard Rink (Indianapolis, Indiana), Emilie Rissman (Charlottesville, Virginia), Paul Saenger (Nueva York, Nueva York), David Sandberg (Buffalo, Nueva York), Justine Schober (Erie, Pensilvania), Norman Spack (Boston, Massachusetts), Barbara Thomas (Rottenburg am Necker, Alemania), Ute Thyen (Lübeck, Alemania), Eric Vilain (Los Angeles, California), Garry Warne (Melbourne, Australia), Jean Wilson (Dallas, Texas), Amy Wisniewski (Des Moines, Iowa), Christopher Woodhouse (Londres, Reino Unido), y Kenneth Zucker (Toronto, Ontario).
Se agradece enormemente el trabajo de Alan Rogol, Joanne Rogol, Pauline Bertrand y Pam Stockham en la organización del encuentro
Traducción: Hortensia Moreno y Eva Alcántara
Revisión técnica: Carlos Alberto Narváez Pichardo
Tomado de Pediatrics, 2006; 118; e488. Versión en línea: http://pediatrics.aappublications.org/content/118/2/e488.fulLhtmI.
DSD ha sido traducido generalmente en la bibliografía médica como TDS (Trastornos del Desarrollo Sexual) y ocasionalmente como dds (Desórdenes del Desarrollo Sexual). En el presente texto utilizamos ambas acepciones de acuerdo a lo ya traducido en tratados médicos especializados [N. de las T.].
Con el asterisco intentamos resaltar la estructura dicotómica que es propia del lenguaje. El idioma español obliga a declinar el género gramatical en las palabras. El uso del asterisco lo retomamos de Mauro Cabral (2009) [N. de las T.].