




El síndrome de hiperinmunoglobulinemia E es una inmunodeficiencia primaria con manifestaciones clínicas multiorgánicas y diversos antecedentes genéticos. La tríada clínica de síntomas observados en la mayor parte de los pacientes con síndrome de hiperinmunoglobulinemia E incluye: abscesos recurrentes de etiología estafilocócica, infecciones respiratorias recurrentes y niveles séricos de inmunoglobulina E elevados (>2.000UI/ml). Se reporta el caso clínico de una mujer con diagnóstico de síndrome de hiperinmunoglobulinemia E a los 4 años de edad, que constituye el primer caso de embarazo a pesar de agenesia ovárica y salpinge, que sobrevive al mismo a pesar de evolucionar con neumonía severa y pulmón con bronquiectasias y neumatoceles gigantes.
Hyperimmunoglobulin E syndrome is a primary immunodeficiency with multiorgan clinical manifestations and various genetic backgrounds. The clinical triad of symptoms observed in most patients with hyperimmunoglobulin E syndrome includes: recurrent staphylococcal abscesses, recurrent respiratory infections, and elevated serum IgE level>2,000IU/ml. We report the case of a woman who was diagnosed with hyperimmunoglobulin E syndrome at 4 years old. This is the first reported case of pregnancy despite unilateral ovarian and Fallopian tube agenesis. The patient survived the pregnancy despite severe pneumonia, bronchiectasis and giant pneumatoceles.
El síndrome de hiperinmunoglobulina E (HIES) es un desorden complejo caracterizado por los niveles de IgE séricos elevados, infecciones estafilocócicas recurrentes graves de piel y pulmón, candidiasis mucocutánea, dermatitis crónica y anomalías esqueléticas y dentales1. Presenta alteraciones en la coordinación de las señales de célula-célula que tienen el potencial de afectar células Th17, células B y respuestas del neutrófilo. Aunque no es un defecto primario de los fagocitos, los neutrófilos de los pacientes con este síndrome presentan un variable y, a veces, profundo defecto quimiotáctico2. Existen 3 formas de presentación del HIES: a) alteraciones en el transductor y activador de la señal de la transcripción 3 (STAT3); b) alteraciones en la señal del dedicador de la citocinesis 8, y c) alteraciones en la señal de la tirosina cinasa 23.
Por otro lado, la agenesia ovárica es un desorden raro que resulta en amenorrea primaria e infertilidad en las mujeres afectadas, con muy pocos casos reportados en la literatura. Pocas alcanzan la pubertad y presentan amenorrea primaria, pérdida de características sexuales secundarias e hipogonadismo hipergonadotrófico. Formas variables de anomalías müllerianas son observadas en todas las pacientes4. Los ginecólogos deben reconocer a estas pacientes de forma temprana, ya que puede existir una morbilidad significativa e incluso mortalidad asociada a esta inmunodeficiencia. Además, las mujeres que llegan a la edad reproductiva deben recibir consejo genético. Este reporte tiene el propósito de difundir el caso clínico de una paciente con HIES, embarazo y agenesia ovárica vista en nuestra institución; además, revisamos la literatura actual.
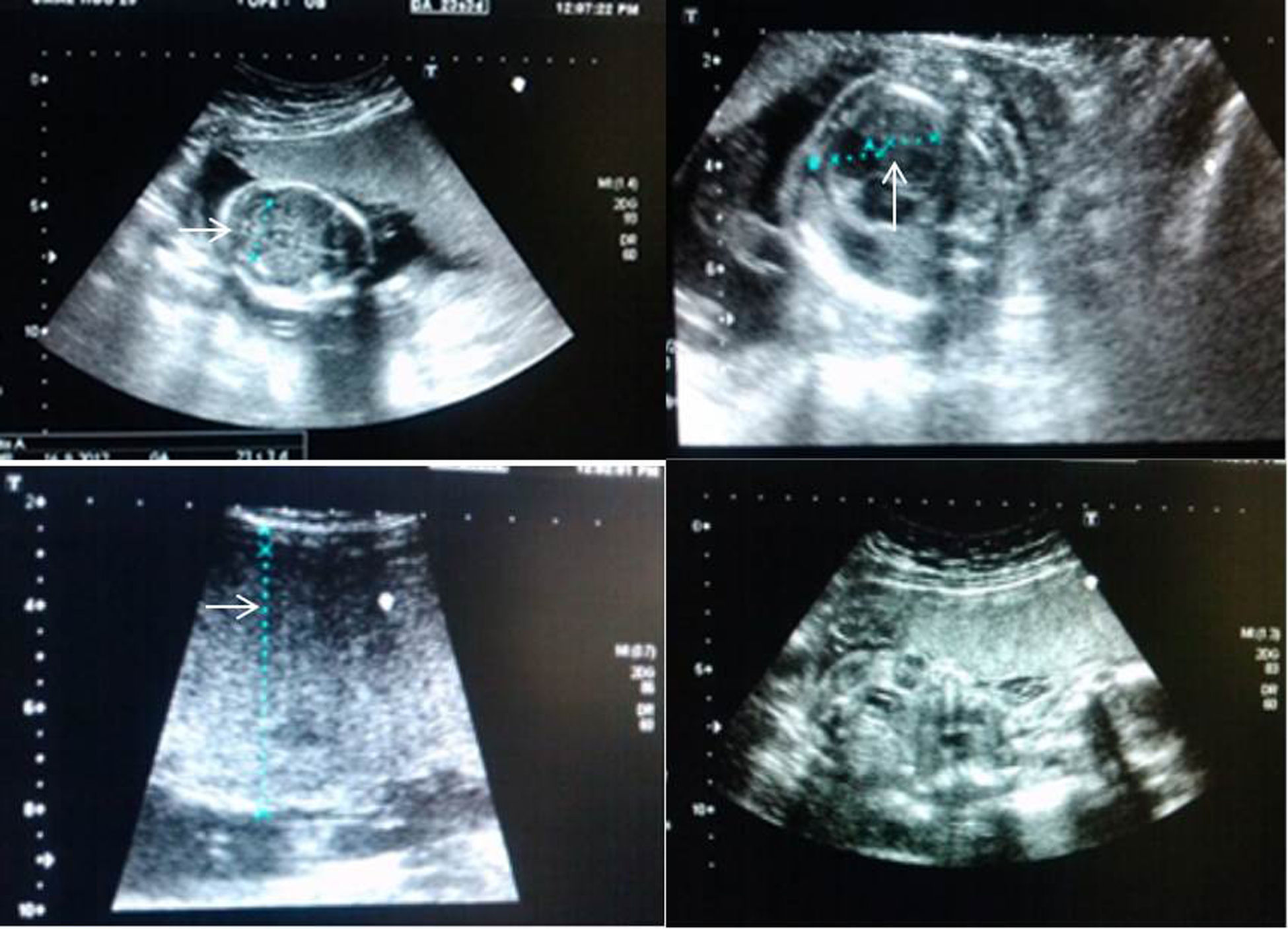
Caso clínicoSe trata de una mujer de 20 años de edad, con 23 semanas de gestación en su primer embarazo. A los 4 años de edad presentó infecciones respiratorias recurrentes severas y se diagnóstico HIES con mutación a nivel de STAT3. A los 11 años se inició tratamiento de inmunoestimulación con extracto dializable de leucocitos, con lo que se logró una disminución de los procesos infecciosos y de los niveles de IgE en los primeros 2 años posteriores al tratamiento. A los 15 años se detectaron bronquiectasias basales bilaterales, con mayor afectación de lóbulo inferior izquierdo. A los 20 años cursó con neumonía severa y se detectó insuficiencia tricuspídea y neumatoceles gigantes en el pulmón derecho. A las 24 semanas de gestación fue revalorada por el Servicio de Medicina Fetal; el ultrasonido obstétrico mostró producto pélvico, con placenta corporal anterior de 38mm, probable placentomegalia, microcefalia, intestino hiperecogénico y áreas hiperecogénicas a nivel hepático con presencia de ascitis (fig. 1); el peso fetal estimado era de 473g. Se realizó cordocentesis y el resultado del análisis para procesos infecciosos fue no concluyente. La citología hemática reportó 12.100 leucocitos/μl (neutrófilos 48%, linfocitos 34%, eosinófilos 13%; cifra total 1.650/μl); la hemoglobina fue de 11g/dl, el hematocrito de 34,5% y las plaquetas de 313.000.

Imagen ultrasonográfica de la parte superior izquierda, corte transcerebelar con hallazgo de hipoplasia de cerebelo. Imagen superior derecha, corte de cuatro cámaras apical donde se observa cardiomegalia e hipertrofia miocárdica. Imagen inferior izquierda, corte longitudinal de placenta con 38 mm de grosor. Imagen inferior derecha, ecografía a las 26 semanas encontrando anhidramnios.
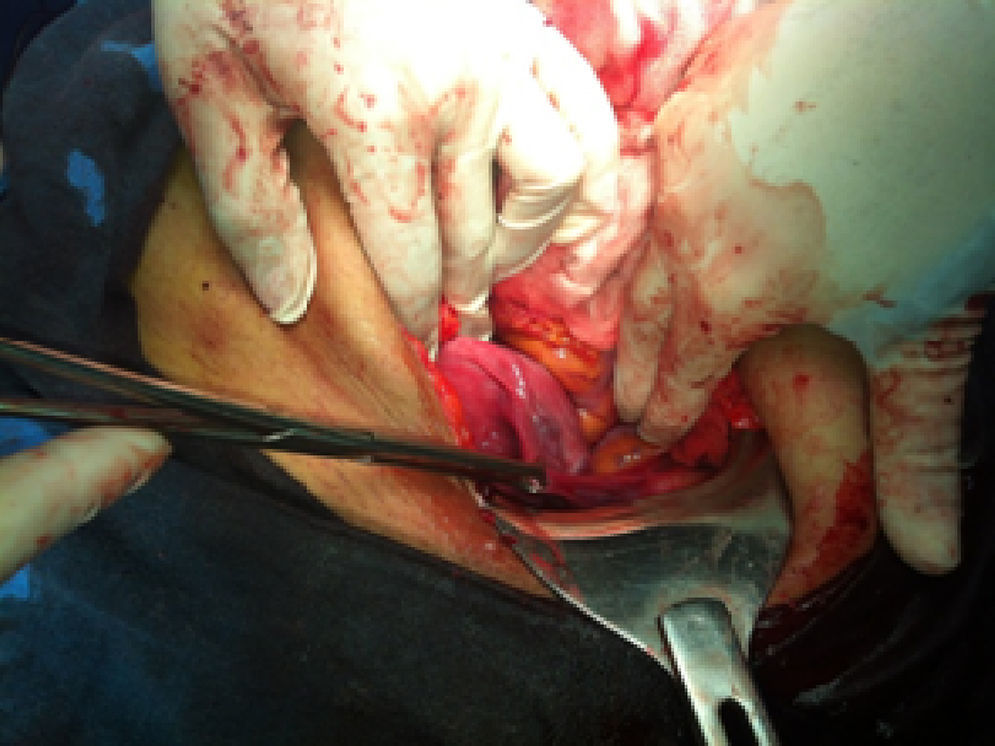
Se convocó al Comité de Ética Médica, el cual aconsejó la interrupción del embarazo por el alto riesgo de las condiciones maternas y del producto; no obstante, la paciente decidió continuar con el embarazo. En la semana 26 de gestación se ingresa para interrupción del embarazo, con anhidramnios, membranas íntegras y un peso fetal estimado de 459g. La fetometría estimó una edad gestacional de 21,6 semanas. Se realizó operación cesárea y se observó la ausencia de líquido amniótico y placentomegalia. Se obtuvo un recién nacido vivo varón de 380g, con una talla de 18cm y Apgar al minuto y a los 5min de 7-7, respectivamente. Como hallazgo se reportó agenesia de ovario y salpinge izquierda, y se realizó salpingectomía bilateral por el riesgo obstétrico alto (fig. 2). El producto falleció a los 2 días del nacimiento. La paciente permaneció por 2 días en la Unidad de Cuidados Intensivos; presentó evolución favorable y se dio de alta sin complicaciones aparentes.
Discusión
Este es el primer caso reportado en México de una paciente con agenesia ovárica y de salpinge unilateral y asociada a HIES. La agenesia de ovario es una alteración rara que se manifiesta con amenorrea primaria, pérdida de desarrollo sexual secundario, hipogonadismo hipergonadotrófico e infertilidad en las mujeres afectadas. Formas variables de anomalías müllerianas son observadas en estas pacientes, como es el caso que se presenta, que se acompañó de ausencia de salpinge ipsilateral4. Se ha mostrado que la incidencia de agenesia unilateral congénita de ovario es de uno por 11.240 casos (0,0089%)5, aunque la estimación de esta anomalía entre la población es bastante confusa debido a que algunos casos pueden pasar inadvertidos y, según nuestro conocimiento, no existen datos de autopsias.
Hay 2 posibles patogenias en relación con la agenesia ovárica unilateral: hipótesis embriológica y mecánica. Un defecto en el desarrollo del sistema mesonéfrico unilateral y mülleriano entero, o un defecto localizado en la región de la cresta genital y la parte caudal del conducto mülleriano se incluirían dentro de la primera razón. Según la hipótesis mecánica, la ausencia de los anexos es debida a la torsión asintomática de la trompa de Falopio y el ovario, con la consiguiente isquemia de órganos y atresia. Esto, eventualmente, puede ocurrir durante la vida adulta, la adolescencia o durante la vida intrauterina6.
La segunda explicación es la separación completa de la gónada de sus accesorios normales. Esta condición puede ser causada por torsión del pedículo del ovario en el nacimiento, infancia o vida adulta7. Hasta la fecha, pocos casos han reportado la agenesia ovárica unilateral asociada a la ausencia total de la salpinge ipsilateral, pero ninguno se ha asociado además al HIES, como sucedió en nuestro caso6,8.
El HIES es una inmunodeficiencia primaria multisistémica caracterizada por eczema, abscesos de la piel, infecciones estafilocócicas recurrentes de la piel y los pulmones, formación de neumatoceles, candidiasis, eosinofilia y niveles séricos de IgE>2.000UI/ml9. También presentan un aspecto facial característico, escoliosis, conservación de los dientes primarios, hiperextensibilidad, fracturas óseas después de trauma mínimo y craneosinostosis10. En 1966, el HIES fue descrito por primera vez en 2 casos de niñas con pelo rojo, dermatitis crónica severa, abscesos fríos y neumonía recurrente, denominando a esta enfermedad síndrome de Job en alusión al personaje bíblico11. Esta enfermedad también fue llamada HIES cuando pacientes similares fueron detectados con niveles elevados de IgE y eosinofilia12.
En los últimos años se han identificado 3 defectos genéticos en HIES. Las mutaciones en STAT3 se asocian a la forma autosómica dominante, mientras que las mutaciones en citocinesis 8, o más raramente en tirosina cinasa 2, se asocian a la forma autosómica recesiva. Recientemente se ha confirmado que la medición de las células Th17 cooperadoras puede ayudar a predecir mutaciones en STAT3. En la forma autosómica recesiva de HIES se encontró que una pérdida de expresión de citocinesis 8 impedía la expansión de células T y la producción de anticuerpos específicos por las células B. Probablemente, estos factores contribuyen a la mayor susceptibilidad a infecciones virales en la piel, las alergias severas y el alto riesgo de malignidades que definen este desorden13–16.
Infecciones pulmonares recurrentes representan un sello clínico en la forma autosómica dominante de HIES. La mayoría de los pacientes tiene al menos un episodio de neumonía, con más del 50% de estos que tienen 3 o más episodios, y el desarrollo de neumatoceles y bronquiectasias afecta hasta a un 75%, como el caso que nos ocupa17.
Los pacientes con HIES tienen una esperanza de vida acortada. La causa principal de muerte en estos pacientes sigue siendo la insuficiencia respiratoria secundaria a las complicaciones de las infecciones pulmonares recurrentes, principalmente por Pseudomonas y Aspergillus spp.18.
Aunque no existe ningún tratamiento curativo, la profilaxis con antibióticos y el tratamiento específico se basan en la atención convencional (por ejemplo, las intervenciones quirúrgicas para el drenaje del absceso), y se aconseja el uso de antibióticos sistémicos en caso de infecciones. Se ha utilizado la terapia de reemplazo de inmunoglobulina y se han probado algunos otros tratamientos, tales como antagonistas de IFN-γ ciclosporina, que parecen ser útiles en el manejo de pacientes con HIES19.
Pocos casos se han reportado de embarazo y HIES, sin embrago, lo que hace más relevante el caso que presentamos es la asociación con agenesia ovárica y salpinge unilateral, ya que estas pacientes suelen ser infértiles20. Además, los hallazgos ultrasonográficos fueron muy indicativos de infección, pero los resultados de laboratorio no fueron concluyentes. Sin embargo, es importante señalar que ante la presencia por separado, pero especialmente en combinación, de hepatomegalia, esplenomegalia, petequias, púrpura, ictericia, microcefalia, encefalopatía, anormalidades oculares, anemia, trombocitopenia, hiperbilirrubinemia o transaminasas hepáticas elevadas, el médico debe considerar la posibilidad de una infección viral congénita21,22.
En los últimos años se han producido espectaculares avances en el conocimiento inmunológico y genético de la enfermedad. Sin embargo, aparecen de forma aislada nuevos datos clínicos que deben ser confirmados en estudios observacionales amplios. Por otra parte, la esperanza de vida ha aumentado en la población occidental, y los ginecólogos deberán orientar a estos pacientes cuando lleguen a la edad adulta y reproductiva; por lo tanto, es importante saber ofrecer un adecuado consejo genético para que la gestación siga un curso normal y tratar de evitar la afectación tanto del producto como de la madre por su predisposición a enfermedades infecciosas23,24.
Conflicto de interesesLas autoras reportan que no existe relación financiera o personal con otras personas u organizaciones que pudieran dar lugar a un conflicto de intereses en relación con el artículo que se remite.
