




De acuerdo con diferentes organizaciones como la Asociación Americana del Corazón o la Organización Mundial de la Salud, las enfermedades cardiovasculares se han convertido en la primera causa de muerte en países occidentales. Aunque la exposición a diferentes factores de riesgo, en particular los relacionados con el estilo de vida, contribuyen de manera significativa a la etiopatogénesis de enfermedades cardíacas, el incremento en la esperanza de vida y el envejecimiento de la población asociado a él se consideran los determinantes principales del inicio y desarrollo de las mismas. Las mitocondrias y el estrés oxidativo se han señalado como factores relevantes tanto en el envejecimiento del corazón como en el desarrollo de enfermedades cardíacas como la insuficiencia cardíaca, la hipertrofia cardíaca y la miocardiopatía diabética. Durante el envejecimiento, diferentes procesos celulares relacionados con la función mitocondrial, como la bioenergética, procesos de apoptosis o de inflamación, se ven alterados, lo que conlleva una reducción en la supervivencia celular, y como consecuencia, disfunción cardíaca. Aumentar nuestro conocimiento sobre los mecanismos mitocondriales relacionados con el proceso de envejecimiento proporcionará nuevas estrategias para mejorar de forma más eficiente este proceso y las diferentes enfermedades relacionadas con él, en particular las cardiovasculares.
According with different international organizations, cardiovascular diseases are becoming the first cause of death in western countries. Although exposure to different risk factors, particularly those related to lifestyle, contribute to the etiopathogenesis of cardiac disorders, the increase in average lifespan and aging are considered major determinants of cardiac diseases events. Mitochondria and oxidative stress have been pointed out as relevant factors both in heart aging and in the development of cardiac diseases such as heart failure, cardiac hypertrophy and diabetic cardiomyopathy. During aging, cellular processes related with mitochondrial function, such as bioenergetics, apoptosis and inflammation are altered leading to cardiac dysfunction. Increasing our knowledge about the mitochondrial mechanisms related with the aging process, will provide new strategies in order to improve this process, particularly the cardiovascular ones.
Según la Organización Mundial de la Salud, las enfermedades cardiovasculares se están convirtiendo en la primera causa de muerte en los países occidentales1, superando a los trastornos neurodegenerativos. En Europa, a pesar de que se ha producido una reducción en los últimos años en el número de muertes relacionadas con las enfermedades cardiovasculares, estas son responsables de la muerte de cerca de 4 millones de personas, casi un 50% de las muertes en toda Europa. Dentro de las estadísticas, España ocupa un lugar destacado entre los países europeos con menor mortalidad debida a enfermedad coronaria, la principal causa de muerte entre las enfermedades cardiovasculares2.
Aunque determinados factores ambientales y relacionados con el estilo de vida como dieta e inactividad física desempeñan un papel importante en la etiopatología de los trastornos cardíacos, el envejecimiento es considerado el principal factor determinante para el desarrollo de enfermedades cardíacas. Por otro lado, otras enfermedades relacionadas con la edad como la dislipidemia y la diabetes mellitus exacerban los efectos negativos del envejecimiento sobre el sistema cardiovascular. De acuerdo con el Instituto Nacional de Estadística, en España, desde 1975, la esperanza de vida ha aumentado cada lustro un año de media. Además, en los últimos 20 años, la esperanza de vida al nacer ha aumentado cerca de 5 años. Este aumento tiene una consecuencia directa: el incremento en la tasa de incidencia de enfermedades relacionadas con la edad, en particular las cardiovasculares. Más importante aún, el aumento de la esperanza de vida media se espera que siga aumentando en los próximos 20 años, cuando cerca del 20% de la población alcanzará los 65 o más años. El corazón es principalmente un tejido posmitótico y tiene un metabolismo altamente aeróbico. Estas características conllevan una alta dependencia de la función mitocondrial para el correcto funcionamiento de las células cardíacas3. Las mitocondrias desempeñan un papel determinante en la función y supervivencia de los cardiomiocitos y son fundamentales para cubrir la alta demanda energética del miocardio. En condiciones fisiológicas, el 20-30% del volumen celular de los cardiomiocitos está ocupado por mitocondrias, pero este valor puede aumentar cuando los requerimientos energéticos del miocardio se incrementan. El corazón consume el equivalente de 6kg de ATP/día, que se genera principalmente a través de la fosforilación oxidativa mitocondrial a partir del catabolismo de lípidos e hidratos de carbono4. El reservorio energético del corazón incluye ATP (≈5μmol/g de peso húmedo) y fosfocreatina (PCr; ≈8μmol/g de peso húmedo), actuando esta última como sistema de transporte y tampón del ATP5. En la mitocondria, el enlace fosfato del ATP puede ser transferido a la creatina por la creatina cinasa mitocondrial para formar PCr. La PCr puede difundir fácilmente a través de la membrana mitocondrial hacia el citosol, donde se puede utilizar para generar ATP a partir de ADP a través de reacciones catalizadas por la creatina cinasa citosólica6. Hay un delicado balance entre la expresión de genes nucleares y mitocondriales, que regula el ensamblaje de complejos respiratorios mitocondriales. Especialmente en condiciones de ejercicio, la biogénesis mitocondrial se activa a través de la modulación de la relación ATP/ADP, la activación de la proteína cinasa activada por AMP, y la consiguiente expresión de los factores PGC-1α y NRF17. A su vez, cuando aumenta la demanda de energía se activa la expresión de genes de ADN nuclear y mitocondrial (ADNmt) maximizándose la capacidad de las mitocondrias para llevar a cabo la fosforilación oxidativa.
Estrés oxidativo e inestabilidad genómica mitocondrialTeniendo en cuenta que el envejecimiento es uno de los principales factores que conducen a la disfunción cardíaca, es especialmente importante conocer los mecanismos que subyacen al proceso de envejecimiento con el fin de encontrar aproximaciones terapéuticas para mejorarlo. Dentro del proceso de envejecimiento, se han considerado relevantes diferentes factores tales como la disfunción mitocondrial, la inestabilidad genómica o las alteraciones epigenéticas, entre otros8. Desde su propuesta como uno de los principales determinantes de la longevidad9, un gran número de estudios han apoyado el papel de las mitocondrias y del estrés oxidativo en el proceso de envejecimiento. Aunque los radicales libres se generan en diferentes compartimentos celulares y por múltiples enzimas, tales como la NADPH-oxidasa o la xantina oxidasa, la producción de especies reactivas de oxígeno (ERO) mitocondriales se considera la fuente más importante de radicales libres celulares en tejidos sanos y condiciones fisiológicas. La razón es que el principal generador de radicales libres, la cadena de transporte de electrones, se encuentra en la membrana mitocondrial interna10. Estos radicales se generan de forma continua durante la fosforilación oxidativa y la generación de ATP. Durante muchas décadas, las investigaciones relacionadas con ERO se centraron en su papel como moléculas oxidantes, sin embargo, estas ERO desempeñan también un papel importante como moléculas de señalización, estando involucradas en diferentes respuestas fisiológicas y en el control de la homeostasis celular11.
En condiciones fisiológicas, una cantidad variable de oxígeno se convierte en el anión superóxido (O2−) debido a la fuga de electrones principalmente de los complejos mitocondriales I y III10,12. La superóxido dismutasa cataliza la reacción que transforma el O2− en H2O2, que es a su vez neutralizado dando lugar a O2 y H2O debido a la actividad de enzimas como la catalasa. Por otro lado, el H2O2 se puede convertir en el radical hidroxilo (OH−) a través de reacciones de Fenton y Haber-Weiss en presencia de hierro. El contenido de hierro mitocondrial aumenta significativamente con el envejecimiento en diferentes tejidos, incluyendo el miocardio, lo que podría estar asociado a un incremento en la generación de OH− y por lo tanto del daño oxidativo en la edad avanzada13,14. La producción continua de O2− y de otras ERO en la mitocondria supone un incremento en el daño oxidativo a las diferentes macromoléculas con la edad. Dentro del daño generado por las ERO, es especialmente relevante el producido sobre el ADN. Debido a dicha exposición continua a radicales libres y a diferentes factores exógenos, la integridad del ADN tanto nuclear como mitocondrial se ve comprometida durante el envejecimiento15,16. En concreto, la inestabilidad del ADNmt se ha considerado especialmente relevante en el proceso de envejecimiento, ya que conlleva disfunción mitocondrial17–19. Los radicales libres generan un gran número de lesiones en el ADNmt, incluyendo bases oxidadas, y roturas simples o dobles de cadena, siendo esta última una de las lesiones del ADN más deletéreas. Muchas de estas lesiones del ADN inducidas por ERO presentan efectos mutagénicos o citotóxicos debido al emparejamiento erróneo de las bases, lo que puede dar lugar a mutaciones tras los procesos de replicación del ADN20,21. El daño continuado sobre el ADN puede provocar el bloqueo de los procesos de replicación y transcripción, generando inestabilidad genómica. A fin de evitar dicha inestabilidad, existen diferentes mecanismos de reparación del ADN, tanto en el núcleo como en las mitocondrias. En función del tipo de daño en el ADN que se haya generado, se activa una ruta particular de reparación22. Sin embargo, a pesar de la existencia de estos mecanismos reparadores, el daño al ADN aumenta y se acumula con la edad. Así, la frecuencia de mutaciones puntuales y deleciones en el ADNmt es aproximadamente 3 veces más alta en el corazón de los ratones viejos que en animales jóvenes23. La importancia de la acumulación de mutaciones en el ADNmt como un factor determinante en el envejecimiento y en la pérdida de función de diferentes tejidos, incluyendo la disfunción cardíaca, se ha confirmado en modelos animales donde existe una mayor inestabilidad del ADNmt. En uno de estos modelos en ratón se impidió la expresión del factor de transcripción mitocondrial A, específicamente en corazón y músculo, dando lugar a un fenotipo con deficiencia de la cadena respiratoria y acumulación de mitocondrias morfológicamente anormales24. Estas alteraciones se asociaban además con una mayor tasa de apoptosis25. Curiosamente, estos animales presentaban una función cardíaca normal al nacer, pero desarrollaban rápidamente miocardiopatía dilatada en el periodo posnatal24. Otro modelo es el ratón polG, desarrollado hace una década26,27. Estos ratones presentan una alta tasa de mutaciones puntuales y deleciones en el ADNmt de todos los tejidos28,29 y se caracterizan por un fenotipo de envejecimiento prematuro. Además, las mitocondrias del corazón de estos ratones muestran altos niveles de carbonilos proteicos, y una actividad de la cadena de transporte electrónico deficiente, con actividad reducida de los diferentes complejos que la constituyen26. También se ha propuesto que los niveles altos de mutaciones puntuales en el ADNmt afectarían al ensamblaje correcto de los supercomplejos mitocondriales, lo que conllevaría la reducción de la producción de ATP y al deterioro bioenergético29. Por otro lado, los ratones polG muestran un incremento en los procesos apoptóticos en el tejido cardíaco y cambios prematuros relacionados con la edad, tales como la hipertrofia cardíaca y una reducción de las funciones sistólica y diastólica23,30. Del mismo modo, cuando estos ratones fueron sometidos a ejercicio físico de resistencia, la aparición de cambios relacionados con la edad, tanto en el músculo esquelético como en el corazón, se retrasó31. El estudio de Safdar et al. indica que ese efecto del ejercicio físico podría estar mediado al menos en parte por la inducción de la biogénesis mitocondrial, la prevención de las mutaciones de ADNmt y la reducción de procesos apoptóticos31. Recientemente se ha desarrollado un nuevo modelo animal que presenta una acumulación aumentada de deleciones de ADNmt en el miocardio32. Como los animales polG, estos ratones acumulan cardiomiocitos con la función mitocondrial alterada, que podría estar relacionado con la arritmia cardíaca prematura que desarrollan.
Una de las principales consecuencias de la inestabilidad del ADNmt es la disfunción mitocondrial que, como se ha mencionado, se cree que contribuye significativamente al proceso de envejecimiento. Pero las mitocondrias no son solo críticas para la obtención de energía celular a través de la fosforilación oxidativa, sino que también participan en un número importante de funciones celulares, incluyendo procesos de apoptosis, la homeostasis del Ca2+ y la señalización redox entre otros. En consecuencia, el deterioro de la función mitocondrial conduce a la pérdida de la homeostasis celular y, por lo tanto, de funcionalidad tisular33,34. Junto a la acumulación de mutaciones en el ADNmt, otros mecanismos contribuyen a la pérdida de función mitocondrial con la edad, tales como la oxidación de proteínas mitocondriales, alteraciones en la composición lipídica de las membranas mitocondriales o alteraciones en los procesos de mitofagia8. Cabe destacar que la función mitocondrial no solo se ve afectada por la pérdida de integridad del ADN de las mitocondrias. Puesto que la mayoría de las proteínas mitocondriales están codificadas por el ADN nuclear, el incremento de la inestabilidad genómica nuclear también contribuye a la alteración de la función mitocondrial35.
Cambios metabólicos en el corazón con el envejecimientoLa principal vía metabólica utilizada por el corazón es la β-oxidación de ácidos grasos de cadena larga que genera un 70-90% del ATP cardíaco36. El ATP restante proviene de la oxidación de glucosa y lactato, así como de pequeñas cantidades de cuerpos cetónicos y ciertos aminoácidos37. El uso de los ácidos grasos o la glucosa como sustrato puede inhibir directamente la utilización del resto de los sustratos (ciclo de Randle). La selección del tipo de sustrato y la interacción entre glucosa y los ácidos grasos utilizados en el corazón se han considerado de gran relevancia en el desarrollo de enfermedades cardíacas, ya que la obtención de ATP a través de la glucosa es más eficiente que a partir de ácidos grasos38. A nivel mitocondrial, los niveles de proteínas implicadas en la respiración mitocondrial y de otras clave en el metabolismo, incluidas aquellas relacionadas con en el metabolismo de ácidos grasos, disminuyen en el corazón con el envejecimiento. Por el contrario, las proteínas implicadas en el metabolismo de la glucosa, así como ciertas proteínas estructurales extracelulares aumentan significativamente con la edad39. Por lo tanto, el aumento de expresión de las proteínas glucolíticas, junto con una disminución de la oxidación de ácidos grasos, indicaría un remodelado metabólico con la edad similar al que ocurre en la insuficiencia cardíaca (IC) en individuos más jóvenes40. Diversas alteraciones relacionadas con el metabolismo mitocondrial se han asociado con el proceso normal de envejecimiento del corazón41,42. Por ejemplo, se ha propuesto que la IC está asociada con un estado hiperadrenérgico reactivo que aumenta la circulación de ácidos grasos libres en plasma, lo que conduce a una alteración del metabolismo de la glucosa y resistencia a la insulina38. Del mismo modo, cambios fisiopatológicos en el metabolismo asociados a la edad, como la diabetes, contribuyen de manera determinante a la acumulación de proteínas oxidadas y de productos finales de glicación/glucosilación avanzada en el corazón43. Más adelante en esta revisión se tratarán algunas de las alteraciones cardíacas desarrolladas con la edad relacionadas con cambios metabólicos.
Remodelado cardíaco en el envejecimientoEl remodelado del miocardio durante el envejecimiento está relacionado con los cambios en la cantidad y la organización de los componentes de la matriz extracelular44. Una disposición adecuada del colágeno impide una elongación excesiva de las fibras, preservando la función cardíaca45. Una de las alteraciones más recurrentes y mejor caracterizadas asociadas a la edad en el corazón es el incremento en la acumulación de colágeno dentro de los cardiomiocitos y alrededor de los vasos sanguíneos46,47. Durante el envejecimiento aumenta la tasa de renovación del colágeno ventricular y su síntesis por parte de los fibroblastos48. La fibrosis asociada a la edad se caracteriza por el aumento en el contenido de colágeno, la disminución en su solubilidad y el aumento del entrecruzamiento de sus fibras49. Junto al acúmulo de colágeno, las modificaciones y el entrecruzamiento de las fibras de colágeno y elastina contribuyen al desarrollo de la calcificación vascular, que aumenta con el envejecimiento50. Otra consecuencia de la matriz extracelular es la esclerosis valvular que afecta a más del 30% de los individuos de edad avanzada. De hecho, el aumento excesivo de fibras de colágeno se asocia con la aparición de rigidez ventricular y disfunción diastólica en el corazón envejecido. Asimismo, en la desestabilización de la matriz extracelular y el remodelado cardíaco la relación entre las metaloproteinasas de la matriz y sus inhibidores tisulares endógenos desempeñan un papel central estimulando la producción de colágeno. Un estudio reciente ha demostrado que en humanos, la degradación de elastina mediada por metaloproteinasas de la matriz contribuye a la mineralización de las válvulas y a su calcificación a través de la estimulación de la acumulación de calcio en las fibras de elastina fragmentadas51. Una de las consecuencias más comunes del remodelado cardíaco con la edad es la hipertrofia ventricular izquierda (HVI) caracterizada por el engrosamiento de las paredes ventriculares. Ensayos clínicos como «The Framingham Heart Study» o «The Baltimore Longitudinal Study on Aging» han demostrado un aumento con la edad de la HVI en adultos sanos sin hipertensión, un factor de riesgo importante para el desarrollo de HVI52. Con la edad van a activarse diferentes mecanismos que favorecen el desarrollo de hipertrofia cardíaca, tales como el estrés mecánico, la pérdida de cardiomiocitos o el ya mencionado aumento de fibrosis intersticial.
Uno de los procesos que favorecen la pérdida de cardiomiocitos es el aumento de la apoptosis durante el envejecimiento. La inhibición de los procesos de autofagia, en parte debidos a la acumulación de lipofuscina en los lisosomas, se considera una de las principales causas del aumento de la apoptosis53. La pérdida de capacidad autofágica provoca la acumulación de componentes celulares dañados y no funcionales, incluyendo orgánulos como las mitocondrias, lo que conlleva la activación de vías apoptóticas54. Por otro lado, cambios en los miocitos también pueden provocar alteraciones en el manejo del Ca2+. El Ca2+ intracelular desempeña un papel crítico en la modulación de la función cardíaca. La liberación de Ca2+ inducida por Ca2+ regula la contractilidad del miocardio a través de la activación de canales iónicos, de la activación del receptor de rianodina y de la actividad de intercambiador de sodio/calcio. La entrada de Ca2+ en el retículo sarcoplásmico por la actividad de la bomba calcio-ATPasa del retículo sarcoplásmico (SERCA2a) conduce a la relajación del miocardio. En estudios recientes se ha demostrado que el aumento del estrés oxidativo observado en el corazón con el envejecimiento también afecta a SERCA2a, disminuyendo su actividad sobre el Ca2+ y prolongando la relajación diastólica55. Además, en un estudio llevado a cabo en ratas hembra Fischer, un modelo animal habitual de IC asociada al envejecimiento56, se observó que las contracciones celulares prolongadas en animales envejecidos producía una disminución del transporte de Ca2+ intracelular y un aumento de la expresión de isoformas β de la cadena pesada de miosina57,58. Estas modificaciones afectan negativamente a la actividad eléctrica y la potencia contráctil de los cardiomiocitos. Boluyt et al. mostraron que se producía una alteración del proceso del acoplamiento-relajación de los miofilamentos dependiente de Ca2+. Del mismo modo, los autores describieron un descenso en la fosforilación de la troponina i en los miofilamentos56.
Alteraciones cardíacas en el envejecimiento. Papel de la mitocondriaDurante el envejecimiento, los procesos fisiológicos decaen progresivamente, disminuyendo el control de la homeostasis y el aumento de la morbilidad. Al mismo tiempo, la incidencia de las enfermedades relacionadas con la edad aumenta. Aunque todos los tejidos se ven afectados, se considera que aquellos que contienen células posmitóticas, tales como el cerebro y el corazón, van a verse especialmente afectados59. El proceso de envejecimiento cardíaco va a suponer diferentes cambios en la fisiología y la bioquímica del corazón y los vasos asociados. Morfológicamente, el corazón sufre un engrosamiento e HVI y del tabique interventricular. Aumenta la rigidez, la cicatrización, la calcificación de las valvas de la válvula aórtica y la esclerosis aórtica. La actividad eléctrica en el miocardio también se ve afectada en el corazón envejecido por calcificación de la válvula mitral y la reducción en el número de células marcapasos, por procesos de apoptosis, en el nodo sinoauricular y auriculoventricular junto con la acumulación de colágeno, tejido adiposo y amiloidosis60,61.
Hipertrofia cardíacaLos cardiomiocitos pueden sufrir alteraciones a través de diferentes procesos como la isquemia, la presencia de sustancias tóxicas, los microorganismos, etc. Estas situaciones patológicas dan lugar a una contracción inadecuada del corazón o a la pérdida de cardiomiocitos, con una respuesta compensatoria que se manifiesta en el remodelado cardíaco que tiene lugar en la hipertrofia cardíaca62. La hipertrofia cardíaca se ha considerado tradicionalmente como una respuesta adaptativa. Durante esta, la síntesis proteica se intensifica, se producen nuevos sarcómeros, los cardiomiocitos se engrosan y se alargan, la pared ventricular se engrosa y aumenta la contractilidad cardíaca. Esta respuesta elimina temporalmente, o al menos reduce, la sobrecarga hemodinámica del corazón63,64. Sin embargo, numerosos estudios han demostrado que la hipertrofia cardíaca se asocia con un riesgo significativamente mayor de desarrollar disfunción diastólica, IC y arritmias malignas65,66. Se ha descrito que la hipertrofia cardíaca patológica está relacionada con el agotamiento de las reservas energéticas, reflejado en niveles de ATP constantes y en una reducción de la proteína C reactiva (PCr)67,68. La relación PCr/ATP disminuye y el ATP se reduce significativamente a medida que la hipertrofia progresa de compensada a IC69. En cultivos de cardiomiocitos, la hipertrofia inducida por angiotensina ii, endotelina 1, norepinefrina, TNFα, o el estrés mecánico se ha asociado al aumento en los niveles de estrés oxidativo y a la activación de diferentes vías de señalización intracelular mediada por ERO, incluyendo al NFkB y proteínas cinasas activadas por mitógenos70. Por otro lado, la HVI producida por sobrecarga de presión se reduce cuando los animales se someten a tratamiento con antioxidantes71.
Insuficiencia cardíacaComo se ha comentado anteriormente, aunque la hipertrofia cardíaca se ha considerado tradicionalmente como una respuesta adaptativa, diferentes estudios han demostrado que se asocia con un mayor riesgo de desarrollo de IC y arritmias malignas65,72,73. La IC es un problema creciente de salud pública, sobre todo debido a la mayor esperanza de vida de la población y su mayor prevalencia con la edad. En los países en desarrollo, en torno al 2% de los adultos padecen IC; se ha observado que la prevalencia aumenta hasta un 6-10% en personas mayores de 65 años74. Los mecanismos implicados en la IC son complejos y multifactoriales. La IC se caracteriza por una reducción en la velocidad de relajación del miocardio, así como por una disminución de la distensibilidad miocárdica4,75. Asimismo, se asocia con disfunción mitocondrial y con un aumento del estrés oxidativo76. Este último, junto a la alteración de las proteínas reguladoras de la homeostasis intracelular del Ca2+ contribuyen a la disfunción cardíaca relacionada con IC66. Además, las alteraciones del manejo del Ca2+ y de la función de los miofilamentos contribuyen a las alteraciones cardíacas. Se ha propuesto que las mitocondrias de las células endoteliales desempeñan un papel importante en la señalización celular como sensores de la concentración local de oxígeno y reguladores de la producción de óxido nítrico77. Asimismo, el aumento de estrés oxidativo afecta directamente a la estructura y a la función de los cardiomiocitos, ya que produce la activación de diferentes vías de señalización implicadas en el remodelado y la IC78. Todo ello indica que existe una relación estrecha entre la producción de ERO, la disfunción mitocondrial y el desarrollo de la IC. A diferencia de la disfunción diastólica en reposo, la función sistólica, valorada a partir de la fracción de eyección, se mantiene con la edad. La IC con fracción de eyección preservada (IC-FEP) no presenta dilatación cardíaca, aunque se caracteriza por un aumento de la presión ventricular, congestión pulmonar, disnea e intolerancia al esfuerzo79. Se ha observado que la incidencia de la IC-FEP se está incrementando en los últimos años80, en particular en mujeres de edad avanzada, donde el 90% de los nuevos casos de IC son IC-FEP81. A pesar de que en principio se consideró que la IC-FEP estaba principalmente causada por disfunción diastólica, estudios más recientes indican que en personas de edad avanzada la IC-FEP se caracteriza por una amplia gama de anomalías tanto cardíacas como no cardíacas. En comparación con la IC con fracción de eyección reducida, el pronóstico global de los pacientes de IC-FEP es similar a aquellos con IC con fracción de eyección reducida aunque con un número de hospitalizaciones mayor en pacientes del primer grupo. La necesidad de estrategias terapéuticas eficaces para la IC-FEP ha impulsado el desarrollo de modelos animales adecuados para el estudio de la IC-FEP. Entre ellos los modelos de constricción aórtica y de hipertensión sistémica se han utilizado ampliamente debido a que la hipertensión contribuye notablemente a la IC-FEP79. Otro ejemplo es la rata Dahl sensible a la sal que se caracteriza por presentar hipersensibilidad a la ingesta de sodio y es probablemente el modelo animal IC-FEP más empleado. Cuando estos animales se alimentan con una dieta rica en sal (8% de NaCl), desarrollan insuficiencia renal, hipertensión (>175mmHg) e HVI desarrollando IC-FEP a las pocas semanas de vida82. La constricción aórtica transversal moderada en animales jóvenes también provoca hipertrofia concéntrica del ventrículo izquierdo compensada, con anomalías muy marcadas en el llenado diastólico. Estas anomalías progresan notablemente representando un buen modelo para el estudio de la IC-FEP.
Miocardiopatía diabéticaEn personas obesas y en aquellas afectadas de diabetes mellitus, la disfunción cardíaca se considera una consecuencia de la miocardiopatía diabética83. La miocardiopatía diabética afecta al miocardio de los pacientes con diabetes y causa un amplio espectro de anormalidades estructurales que llevan a la HVI y a la disfunción sistólica y diastólica, o a una combinación de ambas84. La incidencia de la diabetes se incrementa con la edad, siendo especialmente relevante en el caso de la diabetes tipo 2. La prevalencia global de la diabetes entre los adultos de 60 años o más es del 19%, aproximadamente 135 millones de personas, representando el 35% de todos los casos de diabetes en adultos85. Asimismo, la incidencia de enfermedad coronaria, accidente cerebrovascular, IC congestiva, hipertensión, neuropatía, deficiencia visual y artritis es mayor entre los adultos de edad elevada con diabetes que en los adultos de edad similar sin diabetes86. Podemos diferenciar entre resistencia a la insulina sistémica y cardíaca. La resistencia a la insulina cardíaca se define como la disminución de la sensibilidad a la insulina o la de captación de glucosa estimulada por insulina en el corazón en ausencia de resistencia a la insulina sistémica o factores de riesgo de enfermedad coronaria como obesidad, hiperglucemia, hiperinsulinemia, hipercolesterolemia e hipertensión87. La resistencia a la insulina por sí sola tiene importantes efectos adversos sobre la función cardíaca. De hecho, el comienzo de la hiperglucemia y la diabetes está a menudo precedido por varios años de resistencia a la insulina. Estudios llevados a cabo en ratones knock-out con supresión específica del receptor de la insulina en cardiomiocitos (CIRKO) o del sustrato del receptor de insulina (CIRSKO) han investigado la relación entre resistencia a la insulina y disfunción cardíaca. Los ratones CIRKO y CIRSKO mostraron una reducción en la captación de glucosa estimulada por la insulina así como un deterioro de la función cardíaca. En ratones knock-out para el transportador de glucosa (GLUT4) también se ha observado disfunción cardíaca asociada a resistencia a la insulina88–90. La resistencia a la insulina en presencia de diabetes está asociada con IC. Aproximadamente el 24% de los pacientes totales con IC y el 40% de los pacientes con IC hospitalizados tienen diabetes mellitus y en las próximas décadas se espera que este porcentaje crezca de manera exponencial con el envejecimiento de la población91. La resistencia a la insulina no solo tiene efectos negativos a nivel cardíaco, también aumenta el riesgo de padecer diabetes tipo 2 tanto en hombres como en mujeres de edad avanzada, lo que predispone al desarrollo de enfermedad cardíaca coronaria92,93. Se han propuesto diferentes mecanismos responsables de la disfunción cardíaca inducida por la resistencia a la insulina. Entre ellos, la disfunción endotelial en respuesta a la insulina y la disminución de la longitud de los telómeros de los leucocitos94,95. Dado que las mitocondrias son la principal fuente de ATP para satisfacer las demandas energéticas del corazón, la disfunción mitocondrial se ha considerado como una de las causas subyacentes a las enfermedades cardíacas relacionadas con trastornos metabólicos y la resistencia a insulina96,97. En condiciones fisiológicas normales, el corazón utiliza energía de sustratos, principalmente ácidos grasos e hidratos de carbono, en función de la demanda metabólica y la disponibilidad de los mismos98. Sin embargo, en el contexto de resistencia a la insulina, la capacidad del miocardio para utilizar la glucosa como fuente de energía se reduce38,99. La fisiopatología de la miocardiopatía diabética está muy relacionada con el cambio en la utilización de sustrato100. En la miocardiopatía diabética se ha observado que se produce una acumulación intracelular de intermediarios metabólicos tóxicos, como el acil-CoA de cadena larga y la acilcarnitina, que afectan a la relación mitocondrial ATP/ ADP, disminuyendo la función metabólica mitocondrial101.
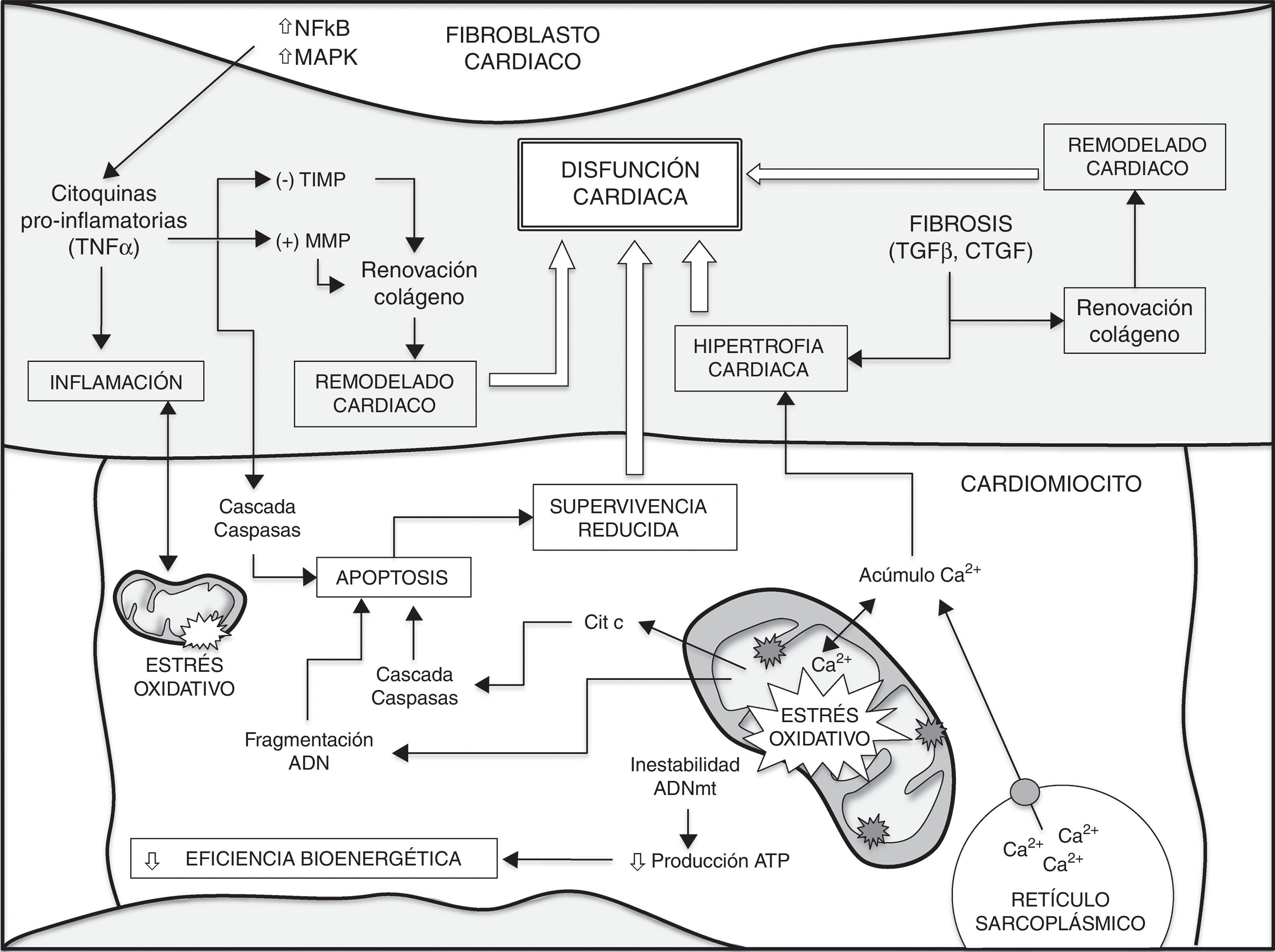
ConclusionesEl desarrollo de numerosas enfermedades cardíacas está asociado con importantes cambios en los procesos metabólicos. Junto a estos cambios, las alteraciones en la biogénesis mitocondrial, así como en el contenido de mitocondrias de los cardiomiocitos y la pérdida de función de estos, contribuyen a producir y agravar un grupo heterogéneo de enfermedades cardíacas. Las ERO de origen mitocondrial, la acumulación de mutaciones en el ADNmt y la pérdida de función mitocondrial desempeñan un papel fundamental en la pérdida de función de los cardiomiocitos y por tanto en el envejecimiento cardíaco. Comprender mejor los mecanismos fundamentales que determinan el proceso del envejecimiento permitirá avanzar en la búsqueda de tratamientos preventivos y terapéuticos para las enfermedades cardíacas. La figura 1 muestra un resumen de los diferentes factores que contribuyen al envejecimiento del corazón y a una mayor susceptibilidad al desarrollo de enfermedades cardíacas.
. El aumento de la inestabilidad del ADNmt, de los procesos apoptóticos, junto a una pérdida de la homeostasis (p. ej., manejo intracelular de Ca2+) en los cardiomiocitos conllevan una menor eficiencia bioenergética y una supervivencia reducida. Por otra parte, la liberación de citocinas a partir de fibroblastos cardíacos y diferentes factores de crecimiento como el TGFβ, desempeñan un papel crítico en la disminución de la supervivencia de cardiomiocitos durante el envejecimiento, en la remodelación cardíaca y en el desarrollo de fibrosis, contribuyendo a la hipertrofia cardíaca. Además, se produce una importante retroalimentación positiva entre el estrés oxidativo y los procesos inflamatorios. Finalmente, todos estos eventos conducen a la disfunción cardíaca. CTGF: factor de crecimiento de tejido conectivo; MAPK: proteínas cinasas activadas por mitógenos; MMP: metaloproteinasas de la matriz; NFκB: factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; TGFβ: factor de crecimiento transformante beta; TIMP: inhibidores tisulares endógenos de las MMP; TNFα: factor de necrosis tumoral alfa.")
Diferentes factores contribuyen al envejecimiento del corazón y a una mayor susceptibilidad a enfermedades cardíacas (ver texto para detalles y referencias).
El aumento de la inestabilidad del ADNmt, de los procesos apoptóticos, junto a una pérdida de la homeostasis (p. ej., manejo intracelular de Ca2+) en los cardiomiocitos conllevan una menor eficiencia bioenergética y una supervivencia reducida. Por otra parte, la liberación de citocinas a partir de fibroblastos cardíacos y diferentes factores de crecimiento como el TGFβ, desempeñan un papel crítico en la disminución de la supervivencia de cardiomiocitos durante el envejecimiento, en la remodelación cardíaca y en el desarrollo de fibrosis, contribuyendo a la hipertrofia cardíaca. Además, se produce una importante retroalimentación positiva entre el estrés oxidativo y los procesos inflamatorios. Finalmente, todos estos eventos conducen a la disfunción cardíaca. CTGF: factor de crecimiento de tejido conectivo; MAPK: proteínas cinasas activadas por mitógenos; MMP: metaloproteinasas de la matriz; NFκB: factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; TGFβ: factor de crecimiento transformante beta; TIMP: inhibidores tisulares endógenos de las MMP; TNFα: factor de necrosis tumoral alfa.
Ambos autores han contribuido equitativamente a este trabajo.
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.