




La enfermedad del hígado graso no alcohólico (EHGNA) comprende una serie de lesiones hepáticas similares a las inducidas por el alcohol, en ausencia de su consumo. Su importancia radica en la alta prevalencia en nuestras sociedades occidentales y, desde el punto de vista hepático, en su progresiva evolución desde esteatosis a cirrosis y cáncer de hígado. Más recientemente, se ha observado que la EHGNA da lugar a frecuentes alteraciones en el metabolismo lipídico y a un incremento del riesgo cardiovascular con aceleración de la arteriosclerosis y de los eventos a ella vinculados. En la presente revisión se hace una actualización de lo publicado hasta la fecha sobre la etiopatogenia de la EHGNA, su influencia en el desarrollo de enfermedades cardiovasculares y las posibilidades terapéuticas vigentes.
Non-alcoholic fatty liver disease (NAFLD) encompasses a series of liver lesions similar to those induced by alcohol but without alchol intake. The importance of this disease lies in its high prevalence in western countries and, from the hepatological point of view, in its progression from steatosis to liver cirrhosis and cancer. More recently, NAFLD has been observed to give rise to frequent alterations in lipid metabolism and an increased cardiovascular risk with acceleration of arteriosclerosis and related events. The present review provides an update on the literature published to date on the etiopathogenesis of NAFLD, its influence on the development of cardiovascular diseases and current therapeutic options.
El término enfermedad del hígado graso no alcohólico (EHGNA) —en la literatura anglosajona «non-alcoholic fatty liver disease» (NAFLD)— se utiliza para denominar a un amplio espectro de afecciones hepáticas, que van desde la esteatosis simple a la esteatohepatitis («non-alcoholic steatohepatitis» [NASH]), y que desde el punto de vista histológico, se asemejan a las lesiones inducidas por el alcohol, si bien por definición, la EHGNA se desarrolla en pacientes que no consumen alcohol o lo hacen en escasa cuantía.
Aunque descrita hace casi 60 años1 y bien caracterizada desde hace treinta2, no es hasta hace poco tiempo en que la EHGNA acapara la atención de los médicos por su alta prevalencia en las sociedades occidentales como la nuestra. Más recientemente se ha observado que la EHGNA no solo puede causar la muerte por el desarrollo de una hepatopatía crónica, sino dar lugar a frecuentes alteraciones en el metabolismo lipídico y a un incremento del riesgo cardiovascular.
En la presente revisión se hace una actualización de lo publicado hasta la fecha sobre la etiopatogenia de la EHGNA, su influencia en el desarrollo de enfermedades cardiovasculares (ECV) y las posibilidades terapéuticas vigentes.
EpidemiologíaLa EHGNA es una enfermedad asintomática, que frecuentemente no muestra alteraciones analíticas relevantes y que en la mayoría de los estudios de prevalencia se determina mediante ecografía. Esta técnica no es excesivamente sensible pues precisa de la infiltración grasa de al menos la tercera parte del parénquima hepático para dar un resultado positivo. Con todas estas limitaciones, se estima que afecta al 20–30% de la población occidental3,4. La esteatohepatitis, que precisa una biopsia hepática para su diagnóstico, afectaría solo al 2–3% de la población5.
Existe una influencia racial en la prevalencia de la EHGNA variando desde un 45% entre los hispanos, a un 24% en afroamericanos y a un 33% en los caucásicos estadounidenses3. Entre los asiáticos acontece en el 25%6. La frecuencia de presentación, al menos en la raza blanca, suele ser el doble entre varones que en mujeres—42% y 24% respectivamente—si bien puede incrementarse en el estado posmenopáusico7.
El predominio de la EHGNA aumenta de forma paralela a la edad: menos del 20% por debajo de los 20 años y más del 40% por encima de los 60. No obstante, también se ha descrito la EHGNA en población infantil con una prevalencia del 2,6%, pero que puede ascender hasta un 10–80% en niños obesos8.
La EHGNA es también más prevalente en pacientes que padecen concomitantemente diabetes mellitus tipo 2 (40–75%), u obesidad (33–76%), llegando hasta el 99% en individuos sometidos a cirugía bariátrica9.
EtiopatogeniaEl hígado desempeña un papel central en el metabolismo lipídico, captando ácidos grasos libres del plasma (AGL), que si no son aprovechados como fuente energética mediante oxidación, son almacenados o exportados tras la síntesis de lípidos y lipoproteínas. Una serie de alteraciones de factores locales y sistémicos, que controlan el equilibrio entre el aflujo, la oxidación y la exportación de lípidos, conduce a la acumulación hepática de triglicéridos. Esta descripción es un resumen de la patogénesis de la EHGNA, que es más compleja, y solo parcialmente conocida, de la que seguidamente se describen los datos más relevantes.
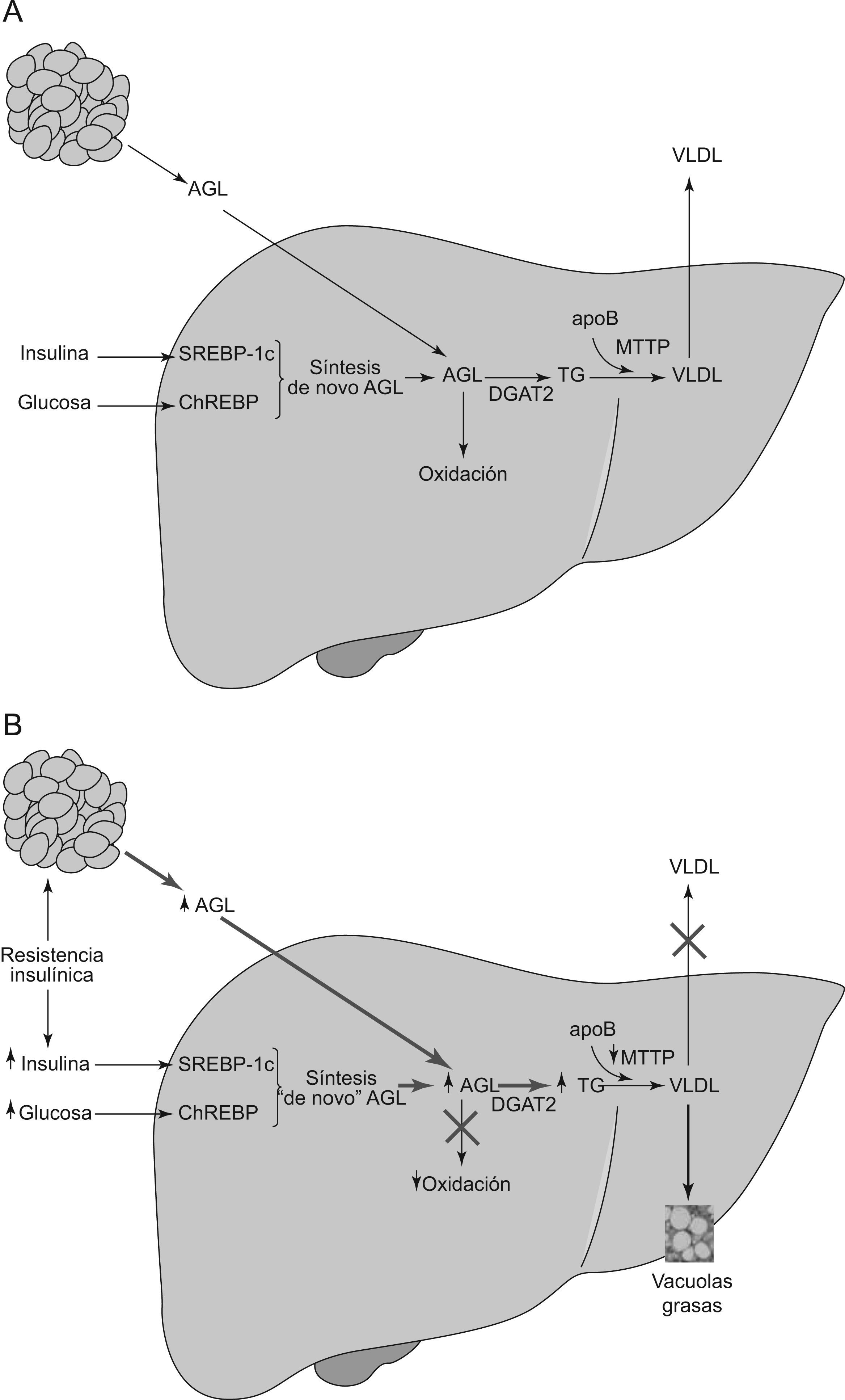
La resistencia a la insulina (RI) y la obesidad, fundamentalmente del tipo visceral, son dos importantes elementos etiopatogénicos de la EHGNA. Ambas aumentan la afluencia de AGL al hígado, lo que induce a una mayor producción de triglicéridos hepáticos. Por otra parte, la hiperinsulinemia y la hiperglucemia, que suelen acompañar a la RI, también pueden promover la lipogénesis «de novo» de AGL, al sobreexpresar factores de transcripción lipogénicos tales como la proteína unida al elemento regulador de esteroles (SREBP-1c) o la proteína unida al elemento de respuesta a los carbohidratos10. Los AGL que no son incorporados a los triglicéridos deben ser metabolizados mediante oxidación en las mitocondrias, peroxisomas y microsomas (fig. 1A). Sin embargo, la activación de SREBP-1c aumenta el malonil-CoA, el cual inhibe la oxidación de los AGL. El resultado neto de estas tres alteraciones es la mayor disponibilidad hepática de AGL como sustrato para la síntesis de triglicéridos11. En esta síntesis, la enzima Acil-CoA:diacilglicerol aciltransferasa (DGAT) juega un papel clave al catalizar el paso final hacia la esterificación de los AGL en triglicéridos. La DGAT está constituida por dos isoformas primarias (DGAT1 y DGAT2). Mientras DGAT1 está presente en varios tejidos, DGAT2 es específica de los hepatocitos. Diversos experimentos en ratones han puesto de manifiesto como su sobreexpresión ocasiona esteatosis, mientras que su inhibición la revierte12,13. Tras su esterificación, los AGL quedan finalmente empaquetados y almacenados como vacuolas de triglicéridos en los hepatocitos, o después de su unión a la apolipoproteína B —mediada por la proteína transferidora de triglicéridos microsomales (MTTP)— son lanzados hacia el torrente sanguíneo en forma de lipoproteínas de muy baja densidad (VLDL). Dado que SREBP-1c inhibe la formación de MTTP, la RI al sobreexpresar SREBP-1c contribuye a una menor incorporación de apoB y a la consiguiente menor formación de VLDL. También puede existir una disminución de la síntesis de apoB14, o una menor respuesta posprandrial de la apoB disociada del incremento de triglicéridos15. En todos los casos, la consecuencia final es la dificultad en el envío del exceso de triglicéridos hacia el plasma. Cuando la tasa de síntesis de triglicéridos sobrepasa la capacidad de producción de VLDL y de su exportación, los triglicéridos se acumulan dentro de los hepatocitos, dando lugar a la esteatosis, marcador definitorio de la EHGNA (fig. 1B).
 Síntesis hepática normal de triglicéridos y VLDL. El contenido en lípidos de hígado viene determinado por el equilibrio de varios procesos: a) Importación de ácidos grasos libres (AGL) del tejido adiposo, b) Síntesis de novo de AGL en hepatocitos, c) Beta-oxidación de los AGL, d) Esterificación de los AGL en TG mediado por la DAGT2 y e) Exportación de triglicéridos como VLDL. Abreviaturas: VLDL: lipoproteínas de muy baja densidad; AGL: ácidos grasos libres; SREBP-1c: proteína unida al elemento regulador de esteroles; ChREBP: proteína unida al elemento de respuesta a los carbohidratos; TG: triglicéridos; VLDL: lipoproteínas de muy baja densidad; Apo B: apolipoproteína B; MTTP: proteína transferidora de triglicéridos microsomales. B) Fisiopatología de la EHGNA. Influencia de la resistencia a la insulina en el EHGNA. La resistencia insulínica y la hiperinsulinemia conducen a la acumulación de lípidos en el hígado por varios mecanismos. En el tejido adiposo, la resistencia a la insulina aumenta la lipólisis de triglicéridos e incrementa los niveles circulantes de los AGL que son captados por el hígado. Además, la hiperinsulinemia y la hiperglucemia aumentan la síntesis de novo de los AGL e inhibe su beta-oxidación. La consecuencia es la acumulación de los AGL dentro de hepatocitos. Para compensarla, la síntesis de TG hepática es impulsada por la sobreexpresión de DGAT2. Por otra parte, la exportación de TG puede verse perjudicada al originarse una disminución de la producción de las VLDL por disminución de la síntesis de la apoB, o de su enlace con los TG, mediada a su vez por una disminución de la MTTP. El resultado neto es un aumento de la concentración intrahepática de TG con su acúmulo dentro de los hepatocitos en forma de vacuolas de grasa, marca constitutiva de la EHGNA. Esta situación inicia un ulterior daño hepático causando una mayor resistencia insulina hepática y la producción de especies reactivas del oxígeno que conlleva el paso desde EHGNA a la esteatohepatitis. Abreviaturas: ↑ = aumentado; ↓ = disminuido; AGL: ácidos grasos libres; SREBP-1c: proteína unida al elemento regulador de esteroles; ChREBP: proteína unida al elemento de respuesta a los carbohidratos; TG: triglicéridos; VLDL: lipoproteínas de muy baja densidad; Apo B: apolipoproteína B; MTTP: proteína transferidora de triglicéridos microsomales; VLDL: lipoproteínas de muy baja densidad.")
A) Síntesis hepática normal de triglicéridos y VLDL. El contenido en lípidos de hígado viene determinado por el equilibrio de varios procesos: a) Importación de ácidos grasos libres (AGL) del tejido adiposo, b) Síntesis de novo de AGL en hepatocitos, c) Beta-oxidación de los AGL, d) Esterificación de los AGL en TG mediado por la DAGT2 y e) Exportación de triglicéridos como VLDL. Abreviaturas: VLDL: lipoproteínas de muy baja densidad; AGL: ácidos grasos libres; SREBP-1c: proteína unida al elemento regulador de esteroles; ChREBP: proteína unida al elemento de respuesta a los carbohidratos; TG: triglicéridos; VLDL: lipoproteínas de muy baja densidad; Apo B: apolipoproteína B; MTTP: proteína transferidora de triglicéridos microsomales. B) Fisiopatología de la EHGNA. Influencia de la resistencia a la insulina en el EHGNA. La resistencia insulínica y la hiperinsulinemia conducen a la acumulación de lípidos en el hígado por varios mecanismos. En el tejido adiposo, la resistencia a la insulina aumenta la lipólisis de triglicéridos e incrementa los niveles circulantes de los AGL que son captados por el hígado. Además, la hiperinsulinemia y la hiperglucemia aumentan la síntesis de novo de los AGL e inhibe su beta-oxidación. La consecuencia es la acumulación de los AGL dentro de hepatocitos. Para compensarla, la síntesis de TG hepática es impulsada por la sobreexpresión de DGAT2. Por otra parte, la exportación de TG puede verse perjudicada al originarse una disminución de la producción de las VLDL por disminución de la síntesis de la apoB, o de su enlace con los TG, mediada a su vez por una disminución de la MTTP. El resultado neto es un aumento de la concentración intrahepática de TG con su acúmulo dentro de los hepatocitos en forma de vacuolas de grasa, marca constitutiva de la EHGNA. Esta situación inicia un ulterior daño hepático causando una mayor resistencia insulina hepática y la producción de especies reactivas del oxígeno que conlleva el paso desde EHGNA a la esteatohepatitis. Abreviaturas: ↑ = aumentado; ↓ = disminuido; AGL: ácidos grasos libres; SREBP-1c: proteína unida al elemento regulador de esteroles; ChREBP: proteína unida al elemento de respuesta a los carbohidratos; TG: triglicéridos; VLDL: lipoproteínas de muy baja densidad; Apo B: apolipoproteína B; MTTP: proteína transferidora de triglicéridos microsomales; VLDL: lipoproteínas de muy baja densidad.
Los AGL pueden hacer progresar la esteatosis hacia la esteatohepatitis por diversos mecanismos, entre los que se incluyen: a) la toxicidad directa; b) el aumento de la síntesis del factor de crecimiento tumoral α (TNFα), a través del factor nuclear kappaB —que a la vez pueden ser inhibidos vía receptor activado de proliferación de peroxisomas tipo gamma (PPARγ) estimulado por los propios AGL—; c) la disminución de la adiponectina —lo que incrementa la RI, la síntesis y depósito de los AGL y los niveles de TNFα—, y por último, d) mediante la generación de especies reactivas del oxígeno (ROS) y la reducción de equivalentes tales como el dinucleótido de nicotinamida adenina (NADH) y la nicotinamida adenina dinucleótido fosfato (NADPH), lo que conlleva a un estrés oxidativo con daño tóxico del hepatocito y a fenómenos de necrosis, proliferación inflamatoria, fibrosis, y muerte celular16.
La teoría de la patogénesis y progresión de la EHGNA mediante «dos golpes» (two-hit hipótesis) está ampliamente aceptada17. El primer golpe sería el acúmulo de triglicéridos en los hepatocitos —esteatosis simple— y el segundo el estrés metabólico-oxidativo y la producción descontrolada de citoquinas que resultan del intento de compensar la alteración de la homeostasis lipídica17. Hay excelentes publicaciones que detallan estos múltiples mecanismos implicados que conllevan a la progresión hacia la esteatohepatitis y la cirrosis, cuya exposición rebasa ampliamente el propósito de esta revisión18–21.
En conclusión, la RI y la obesidad producen una liberación de AGL que estimulan la síntesis y almacenamiento hepático de triglicéridos. Si se sobrepasan los mecanismos de detoxificación hepáticos aparece la lipotoxicidad y la progresión de la EHGNA.
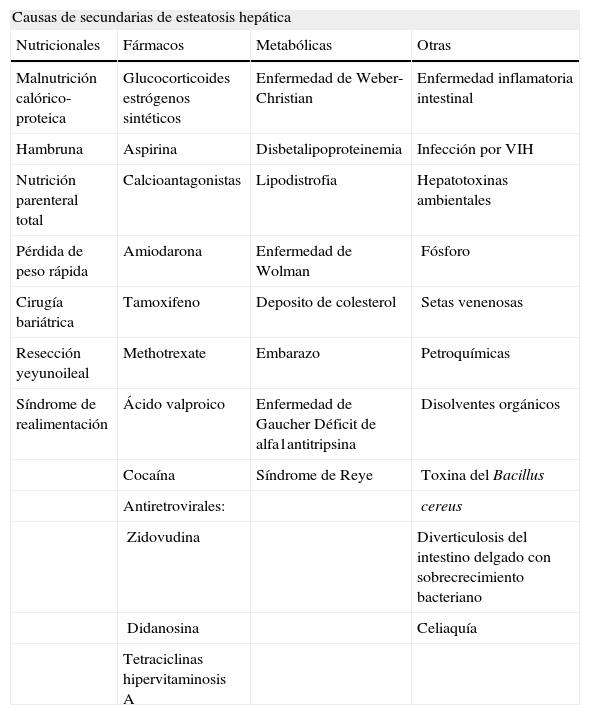
EtiologíaEl principal factor etiológico conocido de la esteatosis hepática es el consumo excesivo de alcohol. Contrariamente y por definición, el consumo de etanol en la EHGNA debe ser mínimo. La mayoría de los grupos que investigan en esta área admiten un consumo máximo de hasta 20g al día en las mujeres y hasta 40g en los varones, cantidades consideradas como potencialmente tóxicas para el hígado. No obstante, hay otras causas secundarias que pueden promover el desarrollo de esteatosis (tabla 1).
Causas secundarias de esteatosis hepática
| Causas de secundarias de esteatosis hepática | |||
| Nutricionales | Fármacos | Metabólicas | Otras |
| Malnutrición calórico-proteica | Glucocorticoides estrógenos sintéticos | Enfermedad de Weber-Christian | Enfermedad inflamatoria intestinal |
| Hambruna | Aspirina | Disbetalipoproteinemia | Infección por VIH |
| Nutrición parenteral total | Calcioantagonistas | Lipodistrofia | Hepatotoxinas ambientales |
| Pérdida de peso rápida | Amiodarona | Enfermedad de Wolman | Fósforo |
| Cirugía bariátrica | Tamoxifeno | Deposito de colesterol | Setas venenosas |
| Resección yeyunoileal | Methotrexate | Embarazo | Petroquímicas |
| Síndrome de realimentación | Ácido valproico | Enfermedad de Gaucher Déficit de alfa1antitripsina | Disolventes orgánicos |
| Cocaína | Síndrome de Reye | Toxina del Bacillus | |
| Antiretrovirales: | cereus | ||
| Zidovudina | Diverticulosis del intestino delgado con sobrecrecimiento bacteriano | ||
| Didanosina | Celiaquía | ||
| Tetraciclinas hipervitaminosis A | |||
La etiología de la EHGNA es probablemente múltiple, estando aún lejos de conocerse todos los agentes participantes. Se admite que influyen factores ambientales y genéticos. Entre los primeros se encuentra el denominado estilo de vida occidental, caracterizado por el binomio dieta abundante y sedentarismo. Un aporte hipercalórico, con exceso de hidratos de carbono, grasa saturada y alimentos con procesamiento industrial, unido a un escaso gasto energético, conducen a la mayoría de los sujetos a una ganancia ponderal. Tanto en población adulta como infantil, el sobrepeso y la obesidad se asocian a la EHGNA. Otro elemento posiblemente implicado es la flora intestinal, que también es un reflejo de la dieta, actuando como facilitador de la permeabilidad intestinal y del paso de endotoxinas22.
Sin embargo, ni todos los pacientes con EHGNA son obesos, ni todas las razas exhiben la misma prevalencia de esta entidad, e incluso, en determinados individuos parece haber un componente familiar23. Estos datos fundamentan la implicación de factores genéticos. Hay evidencias que permiten desvincular la presencia de esteatosis de la obesidad y de la resistencia a la insulina en personas con variantes del gen que codifica la fosfolipasa patatán-like que contiene la proteína 3 (PNPLA3)24. También se ha asociado a la EHGNA con determinados polimorfismos del cambio de un único nucleótido (SNPs) de algunos genes como el de la MTTP25, o el recientemente descrito del gen que codifica la apolipoproteína C3 (apoC3) en asiáticos delgados y sin diabetes26. Un grupo español ha demostrado que los pacientes obesos con EHGNA muestran una sobreexpresión de genes proinflamatorios y proapoptóticos. En aquellos que en la biopsia exhibían fibrosis, se observó, además, una sobreexpresión de genes profibrogénicos, incluyendo el gen del receptor de la leptina27.
HistologíaLos hallazgos anatomopatológicos de la EHGNA son indistinguibles de las lesiones inducidas por el abuso del alcohol. Se exige un mínimo de entre el 5 y el 10% de infiltración grasa para poder definirse como esteatosis hepática28. Además, el amplio espectro de esta enfermedad incluye infiltración de células inflamatorias, balonización de los hepatocitos con presencia en su interior de hialina de Mallory y de acúmulo de gránulos de glucógeno, necrosis de los mismos y fibrosis en el caso de enfermedad avanzada. Para el diagnóstico de esteatohepatitis es necesaria la combinación esteatosis, presencia de polimorfonucleares, monocitos, o ambos, balonización hepatocitaria y áreas de necrosis29. En el 66% de los pacientes con EGHNA se encuentran lesiones mínimas de fibrosis en el momento de su diagnóstico histológico, mientras que en el 25% hay fibrosis septal, y el 15% presenta cirrosis bien establecida. Existe una clasificación clásica de la severidad histológica de la enfermedad, mediante la valoración del porcentaje de infiltración grasa, del grado de inflamación y de los estadios de la fibrosis29. Más recientemente ha visto la luz un nuevo sistema de puntuación semicuantitativo, basado en el anterior, fundamentalmente para unificar criterios a la hora de realizar nuevos ensayos clínicos30.
Historia natural de la enfermedadLos estudios clásicos que llevaron a cabo un seguimiento de pacientes diagnosticados de EHGNA en centros especializados apuntaban hacia un aumento de la mortalidad de estos pacientes31–34. En el último quinquenio se han publicado estudios poblacionales que, además de confirmar una menor supervivencia, han permitido precisar la evolución de esta enfermedad35–38.
Alrededor del 12–40% de los pacientes con infiltración grasa simple desarrollaran esteatohepatitis con fibrosis en un plazo entre 8 y 13 años. De ellos, entre un 15 y un 25% desarrollarán cirrosis en un periodo similar39. La EHGNA también incrementa el riesgo de hepatocarcinoma, presentando una mayor incidencia en la esteatohepatitis40,41. Un 7% de los enfermos cirróticos desarrollarán hepatocarcinoma y el 50% requerirán un transplante o morirán por descompensación de la enfermedad hepática42. La obesidad y la diabetes mellitus muestran una mayor tendencia a la progresión hacia la esteatohepatitis18,43. La EHGNA se asocia con una significativa mayor tasa de mortalidad hepática y eleva un 70% la probabilidad de morir por cualquier causa38. La presencia de esteatohepatitis multiplica por diez el riesgo de cirrosis y por ocho la mortalidad hepática, siendo ambas más probables a medida que aumenta la gravedad del estadiaje de la fibrosis44. La mayoría de los trabajos mencionados con anterioridad proceden de grupos que investigan y publican en el área de hepatología, y aunque en las comunicaciones más recientes se reconoce a las enfermedades cardiovasculares como la causa de mortalidad más frecuente en los pacientes con EHGNA35–38, no se profundiza en su posible implicación en el desarrollo de eventos cardiovasculares.
DiagnósticoNo se ha encontrado ningún biomarcador con las características necesarias que permita un diagnóstico preciso de la EHGNA y que diferencie los estadios leves de los más avanzados de esteatohepatitis y fibrosis45. Por tanto, la biopsia hepática continua siendo el patrón oro del diagnóstico de la EHGNA46. Sin embargo, dado que es un procedimiento invasivo con un 0,5% de complicaciones, que su interpretación es subjetiva y sometida a la variabilidad según la muestra y a la alta prevalencia de la EHGNA, debería limitarse su empleo a los ensayos y estudios clínicos47.
Por otra parte, la mayoría de las veces la EHGNA cursa de forma asintomática o con síntomas y signos inespecíficos – fatiga, dolor en hipocondrio derecho, hepatomegalia… – por lo que en la práctica, los pacientes son diagnosticados por la combinación de niveles elevados de las enzimas hepáticas junto a los hallazgos ecográficos.
La elevación de las transaminasas es la anomalía más usual observada en la EGHNA, estando presente entre el 50 y el 90% de los casos. Si bien cualquier estadio de la enfermedad puede cursar con transaminasas normales, estas suelen estar más elevadas en la esteatohepatitis. Un cociente entre aspartato-aminotransferasa (AST)/alanina-aminotransferasa (ALT) mayor de uno predice la presencia de fibrosis48. La gammaglutamiltranspeptidasa (GGT) y la fosfatasa alcalina pueden estar normales o elevarse hasta el triple de lo normal.
La ecografía es la técnica de cribado preferida para el diagnóstico de la EHGNA en personas con elevación de las transaminasas. Aunque para la detección de la esteatosis la ecografía precisa una infiltración grasa del 30%49, su sensibilidad está en torno al 60%, aumentando hasta un 90–100% en los casos de depósito moderado o severo, con una especificidad del 85% (56–100%)50.
La tomografía computarizada es de menor utilidad diagnóstica que la ecografía en la esteatosis difusa. El diagnóstico se hace cuando el coeficiente de atenuación del hígado en estudios sin contraste es 20 UH menor que la del bazo51. Su precisión puede verse limitada por la carga de hierro hepático. La exposición a la radiación limita aún más su empleo.
La resonancia nuclear magnética (RM) puede ofrecer imágenes y métodos espectroscópicos de cuantificación de la grasa bastante precisos. Las técnicas de gradiente-eco y de potenciación pueden tener utilidad clínica en el diagnóstico de las esteatosis focales. La espectroscopia suele reservarse como herramienta de investigación en estudios epidemiológicos o terapéuticos. La RM-elastografía tiene la capacidad de estimar el grado de fibrosis y parece una herramienta prometedora en la diferenciación entre EHGNA y esteatohepatitis52.
EGHNA y lípidosDerivado de la presencia de RI en la EHGNA, la sobreproducción de VLDL se traduce en un perfil de lípidos séricos de los sujetos con hígado graso caracterizado por niveles elevados de triglicéridos, bajos de colesterol HDL y un aumento en las partículas de LDL pequeñas y densas53–55. La hipertrigliceridemia es la alteración analítica más frecuente. En la población general, el hallazgo de una hipertrigliceridemia o de una dislipemia mixta multiplica por 5,9 y 5,1 respectivamente la posibilidad de infiltración grasa en la ecografía hepática56. Aunque la EHGNA se asocia con la obesidad, preferentemente visceral, los hallazgos lipídicos característicos son independientes de ella57. La presencia de esteatosis hepática en los pacientes con diabetes mellitus tipo 2 agrava la dislipemia diabética, independientemente de la hiperglucemia58. Los niveles de apoA1 son más bajos en la EHGNA, especialmente en aquellos pacientes con fibrosis hepática55. Aunque las concentraciones de apoB están elevadas, ningún grado de esteatosis supone un riesgo de incremento añadido de esta lipoproteína en los diabéticos58. Los niveles de Lp(a) no varían con respecto a la población general53–55.
EGHNA y síndrome metabólicoEl síndrome metabólico (SdMet) esta constituido por una reunión de anormalidades bioquímicas en el que la obesidad visceral y la resistencia insulínica son sus componentes esenciales59. El SdMet se asocia a un incremento del riesgo de diabetes mellitus tipo2 (DM2) y de ECV60. Aproximadamente el 90% de los pacientes con EHGNA tienen más de un componente del SdMet, entre el 35 y el 75% cumple con los criterios diagnósticos y un tercio posee los cinco61. Por ello, comparado con la población general, la prevalencia del SdMet se multiplica entre dos y tres veces en la EHGNA62. Por otra parte, la presencia del SdMet predice un mayor riesgo de desarrollo de la EHGNA en ambos sexos, haciendo improbable la regresión de la infiltración grasa del hígado y facilitando la progresión hacia esteatohepatitis y cirrosis63. Dado que comparten muchos mecanismos fisiopatológicos, se considera a la EHGNA como la representación hepática del SdMet62,64–66.
EHGNA y riesgo cardiovascularLa evidencia actual destierra la vieja idea de que la EHGNA es una enfermedad inocua.
En el apartado referente a la historia natural de la enfermedad se han aportado datos de cómo puede progresar hacia la muerte de causa hepática (cirrosis, hepatocarcinoma o complicaciones de hepatopatía crónica). Aquí se profundizará en la relación de la EHGNA con las ECV.
EHGNA, factores de riesgo y cálculo del riesgo cardiovascularYa hemos visto como la EHGNA se asocia a la RI, al SdMet y a sus componentes, así como a alteraciones lipídicas proaterogénicas. Está bien establecido que los cocientes lipídicos – colesterol total/colesterol- HDL; colesterol-LDL/colesterol-LDL; triglicéridos/colesterol- HDL – son potentes predictores del riesgo cardiovascular (RCV)67,68. Usando la estimación aportada por los cocientes lipídicos se ha determinado no solo que el RCV está aumentado en los pacientes con EHGNA, sino que progresa paralelamente al avance hacia la esteatohepatitis de la enfermedad69. Si el cálculo se determina mediante ecuaciones o tablas de riesgo, nuestro grupo demostró hace tiempo que los pacientes con EHGNA tienen una mayor probabilidad de sufrir eventos cardiovasculares70, lo que ha sido corroborado por series mayores y más recientes71–73. En estas valoraciones, el riesgo también se incrementa con la severidad de la histología hepática72. Diversos estudios han comprobado como muchos de los nuevos factores o marcadores de riesgo – PCR, LDL oxidada, IL6, PAI-1 – se asocian también a la EHGNA74. Además, el riesgo de progresión a diabetes – que asociada a la EHGNA podría considerarse un equivalente coronario – se eleva en esta hepatopatía, de tal forma que a los cinco años entre el 20 y el 25% de los pacientes con EHGNA debutarán como diabéticos35,75,76. En la esteatohepatitis la probabilidad es mayor36. Una vez establecida la DM2, la presencia de la EHGNA predice un aumento de las complicaciones microvasculares como la retinopatía y la nefropatía77.
EHGNA y marcadores subrogados de la arteriosclerosisLa EHGNA ocasiona en ambos sexos un incremento precoz del grosor íntimo-medial carotídeo y una mayor prevalencia de placas tal y como fue demostrado por primera vez por nuestro grupo en pacientes de ambos sexos78. Otros estudios de cohortes, así como poblacionales, y un metanálisis han corroborado estos hallazgos79–83. También se ha observado mediante angiografía coronaria una mayor prevalencia y severidad de las lesiones coronarias en pacientes con esteatosis hepática84,85.
Se ha comprobado que los pacientes con EHGNA sufren además una disfunción endotelial – evaluada tanto por vasodilatación mediada por el flujo de la arteria braquial, como por presencia de moléculas de adhesión – que no depende de la obesidad, ni de la presencia del SdMet o de cualquiera de sus componentes86,87. Otras técnicas de detección de RCV en pacientes asintomáticos han mostrado una mayor prevalencia de anomalías en la EHGNA88,89.
EHGNA e incremento de la prevalencia de enfermedades cardiovascularesAunque de los estudios de prevalencia no se puede obtener relaciones causa-efecto, orientan hacia la posible interacción entre patologías concomitantes. Así, un estudio de trabajadores en Taiwán reveló que aquellos que exhibían una ecografía compatible con esteatosis hepática tenían mayor prevalencia de cardiopatía isquémica, independientemente de la obesidad y otros factores pronósticos90. Una recopilación de publicaciones de pacientes con estatohepatitis muestra cómo los que la padecen viven menos, siendo la enfermedad coronaria la principal causa de muerte91. También se ha comprobado que los pacientes diabéticos con EHGNA tienen más prevalencia de coronariopatía, de enfermedad cerebrovascular y de arteriopatía periférica que los que no la padecían, independientemente del SdMet y de sus elementos constituyentes92,93. Por último, en una serie autópsica infantil, los niños que padecían de hígado graso doblaban la frecuencia de presentación de lesiones coronarias94.
EHGNA e incremento de la incidencia de enfermedades cardiovascularesCada vez en mayor número, una serie de estudios muestran una superior incidencia de morbimortalidad en pacientes con EHGNA, independientemente del método diagnóstico de la misma. Así, autores que han empleado la elevación de la ALT o de la GGT como marcadores subrogados de la EHGNA y que han seguido extensas poblaciones durante períodos entre 10 y 19 años, encuentran que los pacientes con enzimas hepáticas elevadas presentan, con respecto a los que tienen niveles normales, un incremento tanto de las ECV como de la mortalidad debida a ellas, siendo la mayoría de las veces independientes de otros factores tradicionales, incluidos la edad o el peso95–101.
Similares resultados obtienen los estudios en que se definió la presencia de EHGNA mediante ecografía hepática, con un tiempo de seguimiento entre 5 y 12 años102–105.
Por último, como se comentó anteriormente, el empleo de la biopsia hepática hace indiscutible la precisión del diagnóstico de la EHGNA. Pues bien, todas las series que emplean este procedimiento31,34–37,106,107–menos una108– encuentran en estos pacientes, tras un plazo de observación entre 7,6 y 18 años, una disminución de la supervivencia, cuya principal causa fue la mayor incidencia de eventos cardiovasculares y de la mortalidad asociada a ellos. Entre las anteriores publicaciones, los estudios basados en cohortes poblacionales34–36 permiten definir la relevancia de la morbimortalidad de causa arteriosclerótica como un constituyente que acorta la esperanza de vida en aquellos que padecen esta enfermedad, inclusive en los pacientes diabéticos107.
En conjunto, todos estos resultados confirman la hipótesis de que los pacientes con EHGNA tienen un alto riesgo de padecer, o incluso de morir por una enfermedad cardiovascular. Por tanto, la detección casual de una esteatosis hepática en un examen ecográfico convencional debería alertar a los clínicos sobre la probable coexistencia de múltiples factores de riesgo cardiovascular subyacentes que deben ser investigados y tratados, a la par que se debe mantener una vigilancia del posible avance de la enfermedad hepática77,109.
TratamientoAunque la RI es el principal factor patogénico de la EHGNA, otros muchos influyen en su desarrollo. Por tanto, los posibles objetivos terapéuticos son múltiples. Algunas de las actuaciones para conseguirlos comparten la mejoría de las alteraciones histológicas hepáticas con la disminución del riesgo vascular. Nos centraremos aquí principalmente en aquellos fármacos empleados en el tratamiento de los factores de riesgo aunque se repasarán algunas otras terapias ensayadas para revertir los cambios que configuran esta enfermedad.
Modificaciones del estilo de vida como tratamiento de la EHGNAPérdida de peso y ejercicioLa intervención dietética y la práctica de ejercicio constituyen la piedra angular del tratamiento de la EGHNA. Su eficacia queda limitada por el cumplimiento del paciente. Aún así, la pérdida de peso inducida por ambos se asocia a cambios en la fisiopatología que conducen a una mejora de la sensibilidad a la insulina, a una disminución del aporte hepático de AGL y a una reducción de los mecanismos inflamatorios. Sin embargo, carecemos de largos estudios prospectivos y controlados que establezcan una evidencia irrefutable sobre el beneficio histológico de estas medidas. De las aportaciones disponibles, se infiere que hasta una modesta reducción ponderal del 10% produciría una mejora de la resistencia a la insulina, de los niveles de transaminasas, de la infiltración grasa y de los fenómenos inflamatorios y fibróticos110–113.
No se ha analizado cuál debiera ser la composición cualitativa de la dieta en la EHGNA. Únicamente parece que una dieta baja en grasa reduciría el depósito lipídico hepático114 y que los suplementos de ácidos grasos omega 3 mejoran la RI, así como los niveles de triglicéridos y de transaminasas en pacientes con esteatosis115.
Los datos con respecto a la posibilidad de tomar alcohol en el EHGNA son contradictorios. Mientras algunos propugnan que su empleo a dosis bajas puede proteger del desarrollo de la enfermedad116, otros desaconsejan su consumo una vez establecida117,118.
Cirugía bariátricaLa cirugía bariátrica ha mostrado igualmente una mejora de los componentes del SdMet con una drástica reducción de la esteatosis hepática y una disminución del estadio de la EHGNA119,120. No obstante, si la dieta o la cirugía conllevan a una pérdida ponderal excesivamente rápida – mayor de 1,6kg/sem – pueden agravar la esteatohepatitis y la fibrosis121.
Puesto que la dieta, el ejercicio y la disminución de peso tienen un papel firmemente establecido en la mejora del perfil lipídico, de los componentes del SdMet y de la morbimortalidad cardiovascular, su implementación en los pacientes con EHGNA mejorará los dos procesos por los que su supervivencia se ve amenazada. Además, siendo la EHGNA tan prevalente en nuestra población, la favorable relación costo-efectividad de las modificaciones del estilo de vida se ve multiplicada.
Tratamiento farmacológico de la EHGNADada la ausencia de estudios farmacológicos prospectivos y controlados que demuestren la capacidad de reducir las lesiones hepáticas o mejorar la morbilidad de la EHGNA más allá de la reducción del peso, ningún fármaco posee la indicación establecida para el tratamiento de dicha entidad. No obstante, se repasan los posibles efectos beneficiosos de algunos de ellos.
Fármacos contra la obesidadUna vez desaparecidos la sibutramina y el rimonabant del arsenal terapéutico debido a sus efectos secundarios, solo permanece vigente el orlistat. Sin embargo, este reductor ponderal solo se ha ensayado en series preliminares de pocos pacientes y corta duración, mostrando alguna mejoría en los niveles de transaminasas y en la histología, sin poderse descartar que no sea debido a la disminución de peso122,123.
Sensibilizadores de la insulina y afinesMetforminaEsta biguanida ha demostrado su utilidad tanto en el tratamiento de la diabetes como en el enlentecimiento de la progresión de la intolerancia hidrocarbonada. Su mecanismo de acción se basa en la disminución de la producción de glucosa por el hígado y el aumento de su utilización por el tejido muscular. Diversos estudios con afectos de EHGNA diagnosticados mediante biopsia han puesto de manifiesto que su empleo se asocia a una disminución de las transaminasas, del volumen hepático ecográfico y de una mejora de la infiltración grasa y de los fenómenos necroinflamatorios y de fibrosis124–127. No obstante, los resultados no han sido uniformes. Sería necesaria la práctica de estudios aleatorizados para garantizar su verdadera eficacia.
GlitazonasActúan como agonistas de los PPARγ aumentando la oxidación de los AGL y disminuyendo su síntesis en los hepatocitos, lo que les hace buenos candidatos para el tratamiento de la EHGNA. Los resultados de su empleo han sido dispares, pues en estudios cortos empleando fundamentalmente pioglitazona se ha visto mejoría analítica e histológica128–130. Por el contrario, en un estudio de un año de tratamiento, la rosiglitazona, aunque disminuía los niveles de transaminasas y la esteatosis, no logró reducir la inflamación ni la fibrosis131. Un seguimiento de parte de los pacientes de esta serie tratados durante dos años no constató ninguna mejoría más allá de las obtenidas en el primer año, que se mantuvieron132. Por otra parte, los efectos de las glitazonas tienden a desaparecer tras su retirada. En vista de la insuficiente evidencia del beneficio cardiovascular o hepático de estos fármacos, su empleo como terapia en la EHGNA no está recomendado, aunque tienen un potencial terapéutico que debería ser explorado133.
GlinidasEstos secretagogos de insulina han demostrado en ratones mejorar la RI y la esteatosis. En humanos, un estudio piloto ha mostrado capacidad de mejorar los hallazgos histológicos134.
IncretinasEnsayos preliminares en animales con estos nuevos fármacos y algunos casos comunicados en humanos apuntan hacia una mejora de las transaminasas y de la esteatosis, necesitándose estudios prospectivos que establezcan su utilidad en la EHGNA135.
HipolipemiantesEstatinasLos inhibidores de HGM-CoA reductasa tienen un papel bien establecido en la prevención cardiovascular. Además pueden disminuir las concentraciones de transaminasas y de TNFα en los pacientes con EHGNA. Consiguen mejorar el grado histológico de inflamación, pero no la fibrosis136. Estos efectos son independientes de su poder hipolipemiante. Pese a su posible hepatotoxicidad, no existe riesgo hepático en el empleo de las estatinas en pacientes con EHGNA, inclusive con elevaciones basales discretas de transaminasas137,138.
FibratosLos fibratos actúan sobre los PPARα regulando el metabolismo lipídico intrahepático, disminuyendo la RI y mejorando el perfil lipídico. Pese a ello, ni el clofibrato ni el fenofibrato parece que mejoren las alteraciones analíticas o histológicas propias de la EHGNA139. Un estudio con pocos pacientes utilizando gemfibrocilo refirió una disminución de las transaminasas y de la GGT140.
EzetimibaEste inhibidor de la absorción del colesterol, a través del receptor NPC1L1, ha demostrado en ratones obesos que puede aumentar la sensibilidad a la insulina. En humanos, dos estudios muy recientes parecen indicar una mejoría de los enzimas hepáticos, si bien uno de ellos no encontró cambios relevantes en la ecografía141, el otro describe una mejora en los fenómenos inflamatorios142.
Vitaminas, antioxidantes y otros fármacosVitamina EComo antioxidante, una serie de estudios preliminares «in vitro» e «in vivo» presagiaban la posibilidad de que la vitamina E fuera útil para mejorar los hallazgos de la EHGNA. Sin embargo, los ensayos realizados en humanos143, incluido uno controlado con placebo, de 45 pacientes biopsiados a los que se les prescribió vitamina E y C144, no probaron mejorar los parámetros bioquímicos o histológicos de esta enfermedad. No obstante, una serie publicada recientemente con casi 250 pacientes afectos de EGHNA evidencia una mejora de las transaminasas, de la esteatosis y de la inflamación145.
BetaínaUn estudio piloto de 10 pacientes con EHGNA ha demostrado mejorías bioquímica e histológica de los daños hepáticos tras su tratamiento durante un año con betaína146. Realizado hace casi diez años, aún no se ha llevado a cabo un estudio prospectivo que definiera sus autenticas posibilidades terapéuticas.
Ácido ursodeoxicólico (UDCA)Este ácido biliar natural, sin efectos secundarios relevantes, empleado en el tratamiento de la colangitis esclerosante y de la cirrosis biliar primaria mostró en ensayos con animales su posible eficacia en el tratamiento de la EHGNA. Lamentablemente, el único estudio multicéntrico amplio que trató 107 pacientes con UDCA o placebo durante 2 años no mostró diferencias en la histología entre ambos grupos147.
Otros posibles fármacosUna serie de fármacos han mostrado en animales de experimentación propiedades interesantes que les sitúa como candidatos a ensayos controlados como agentes terapéuticos de la EHGNA. Entre ellos, cabe mencionar a los antagonistas de los receptores de la angiotensina ii, que parecen disminuir la fibrosis hepática148, a la N-acetilcisteína como antioxidante y por último, a la pentoxifilina – inhibidor del TNFα – , al cilostazol – que actúa como antioxidante y elevador de los niveles de adiponectina – y al probiótico VSL#3, ya que los tres mejoran las enzimas y la histología hepáticas149–151.
ConclusiónLa EHGNA es una enfermedad prevalente y potencialmente grave que habitualmente se subestima. Su asociación con el desarrollo de una enfermedad hepática progresiva - que puede llevar a la insuficiencia y cáncer hepáticos - está bien establecida. Más recientemente, como se ha expuesto en la presente revisión, ha sido reconocida su asociación con las ECV y su capacidad para acelerar el progreso de la arteriosclerosis y de la morbimortalidad cardiovascular. Con esta información, centrarse solo en el componente patológico hepático de la EHGNA puede ser una actitud simplemente miópica133. La detección casual de una esteatosis hepática en un examen ecográfico convencional debería alertar a los clínicos sobre la probable coexistencia de múltiples factores de riesgo cardiovascular subyacentes que deben ser investigados y tratados. Aunque existe un creciente cuerpo de evidencia que sugiere que el tratamiento de la EHGNA podría reducir el riesgo cardiovascular, son pocos estudios actuales que han abordado este tema. Se deben realizar estudios que investiguen el impacto de la EHGNA sobre la arteriosclerosis y sus complicaciones así como ensayos clínicos que evalúen nuevos abordajes terapéuticos.