





La poliposis adenomatosa familiar (PAF) es un trastorno hereditario caracterizado por pólipos colorrectales múltiples que presenta un riesgo casi inevitable de cáncer colorrectal (CCR) en los pacientes sin tratar.
ObjetivosEvaluar las características clínicas relacionadas con el riesgo de CCR en el momento del diagnóstico.
Material y métodosSe examinaron los expedientes clínicos de 88 pacientes para recopilar información sobre la edad, la historia familiar, los síntomas, la gravedad de la poliposis y la asociación con el CCR.
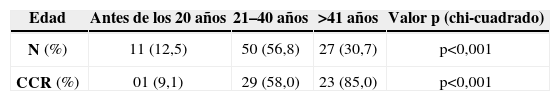
ResultadosSe atendió a 41 hombres (46,6%) y 47 mujeres (53,4%). Se detectó CCR en 53 pacientes (60,2%), con una frecuencia del 9,1% entre aquellos menores de 20 años, 58% entre 21 y 40 años y 85% entre los mayores de 41 años. En el momento del tratamiento, la edad media de los pacientes sin CCR era inferior (29,5 años frente a 40,0 años; p=0,001). Un total de 58 pacientes (65,9%) notificó la existencia de antecedentes familiares, y la edad media de estos no era diferente de los que no notificaron dicha condición (33,4 frente a 34,4 años; p=0,17). Los pacientes asintomáticos representaban el 10,2% del total; en este grupo, la incidencia de CCR fue mucho menor comparada con la que presentaban síntomas (1,1% frente al 65,8%; p=0,001). En los pacientes sin CCR la duración de los síntomas era más corta (15,2 frente a 26,4 meses; p=0,03), y la pérdida de peso menos frecuente (11,4% frente al 33,9%; p=0,01). En la colonoscopia, la poliposis se clasificó como atenuada en 12 pacientes (14,3%), que presentaban una edad promedio superior (48,2 frente a 33,3 años; p=0,02) y una incidencia de CCR idéntica (58,3% frente a 58,3%; p=0,6), comparados con aquellos con poliposis clásica.
ConclusionesEl riesgo de CCR en los pacientes con PAF 1) aumenta de forma significativa después de los 20 años de edad; 2) se asocia con una edad mayor así como una pérdida de peso, presencia y duración de la sintomatología superiores, y 3) es similar en los pacientes con fenotipo atenuado y clásico.
Familial Adenomatous Polyposis (FAP) is a hereditary disorder with multiple colorectal polyps that exhibit an almost inevitable risk of colorectal cancer (CRC) in untreated patients.
GoalsTo evaluate clinical features related to CRC risk at diagnosis.
Material and methodsCharts from 88 patients were reviewed to collect information regarding age, family history, symptoms, polyposis severity and association with CRC.
Results41 men (46.6%) and 47 women (53.4%) were assisted. CRC was detected in 53 patients (60.2%), with a frequency of 9.1% under 20 years, 58% between 21–40 and 85% over 41 years of age. Average age of patients without CRC was lower at treatment (29.5 vs. 40.0 years; p=0.001). Family history was reported by 58 patients (65.9%), whose average age did not differ from those who didn’t report it (33.4 vs. 34.4; p=0.17). Asymptomatic patients comprised 10.2% of the total; in this group, CRC incidence was much lower when compared to those presenting symptoms (1.1% vs. 65.8%; p=0.001). Patients without CRC presented a shorter length of symptoms (15.2 vs. 26.4 months; p=0.03) and less frequent weight loss (11.4% vs. 33.9%; p=0.01). At colonoscopy, polyposis was classified as attenuated in 12 patients (14.3%), who presented greater average age (48.2 vs. 33.3 years; p=0.02) and equal CRC incidence (58.3% vs. 58.3%; p=0.6) when compared to those with classic polyposis.
ConclusionsThe risk of CRC in FAP patients 1) increases significantly after the second decade; 2) is associated with higher age, weight loss, presence and duration of simptomatology; 3) is similar in patients with attenuated or classic phenotype.
La poliposis adenomatosa familiar (PAF) es una enfermedad genética de carácter dominante causada por una mutación en el gen APC («poliposis adenomatosa coli»)1 que predispone al desarrollo de numerosos pólipos colorrectales adenomatosos. Según los datos procedentes de registros nacionales, este trastorno comprende menos del 1% de los casos de cáncer colorrectal (CCR), y afecta de igual manera a ambos sexos: un caso para cada 6–22mil recién nacidos2–4. Aunque sumamente penetrante, hasta un tercio de los pacientes puede que no presente antecedentes familiares algunos, incluidos aquellos con mutación de novo5,6.
La naturaleza adenomatosa y el gran número de pólipos son responsables del riesgo de neoplasia maligna en pacientes sin tratar, con posibilidad de desarrollar cáncer colorrectal (CCR) entre los 10 y los 15 años después de la aparición de los pólipos (unos 35 años), y riesgo de mortalidad en los primeros años de la cuarta década de vida7,8. Por este motivo, los pacientes ser sometido a una colectomía profiláctica para evitar el desarrollo de CCR, que es la causa principal de muerte en este grupo.
Durante los años iniciales, los síntomas clínicos son leves o incluso ausentes. El diagnóstico de la PAF clásica se basa en la detección de más de 100 pólipos colorrectales adenomatosos pequeños durante la pubertad9. Sin embargo, los pacientes con la forma atenuada de la enfermedad presentan un número más reducido de lesiones, que son generalmente planas y ubicadas en los segmentos proximales del colon. En este grupo, el CCR se desarrolla más tarde, sobre los 50 años de edad10–12. Recientemente, se ha descrito otro gen (MUTYH) que está asociado con una forma atenuada de poliposis 13–15.
Además del intestino grueso, la PAF se considera actualmente una enfermedad sistémica que puede implicar a las tres capas germinativas y causar diferentes manifestaciones extraintestinales en el ectodermo (quistes en piel, lesiones retinianas, tumores endocrinos, tumores en el sistema nervioso central), el endodermo (tumor hepático, adenomas gástricos y en el intestino delgado, así como adenomas y adenocarcinomas biliares), y en el mesodermo (anormalidades dentales, tumor desmoide y osteomas)16,17.
Durante las últimas décadas, hemos intentado recopilar datos clínicos, operatorios, endoscópicos e histológicos acerca de la PAF en nuestra unidad de cirugía colorrectal, en un intento de añadir información a los artículos anteriormente publicados por nuestro equipo16,18–24 y documentar las características de esta enfermedad en Brasil.
Por lo tanto, el objetivo del presente trabajo fue evaluar las principales características clínicas de los pacientes con PAF que podrían asociarse con riesgo de CCR en el momento de diagnóstico.
Material y métodosEste estudio fue aprobado por el Comité de Ética de nuestro hospital.
Características de pacientes y datosLa población constaba de pacientes con PAF operados en la Unidad Colorrectal (Universidad de São Paulo) desde 1977 a 2006. Los datos procedentes de los expedientes clínicos se recopilaron retrospectivamente e incluían datos clínicos (sexo, edad, historia familiar, síntomas, exploración física), endoscópicos (distribución de adenomas, gravedad de la poliposis, presencia de CCR, localización topográfica, tumores sincrónicos y los datos operatorios e histológicos (asociación con CCR).
La gravedad de la poliposis se valoró en conformidad con la opinión del colonoscopista con respecto al número y distribución de lesiones en todo el colon y recto. Se clasificó como severa o intensa, cuando el endoscopista usó palabras como «numerosos», «miles» o «mucosa como una alfombra» y como leve o atenuada, cuando la descripción incluía términos como «pocos», «dispersos», «raros» o cuando había menos de 100 adenomas en todo el colon y recto.
Análisis estadísticoGran parte de la información recopilada se utilizó para comparar a los pacientes con y sin CCR. El análisis estadístico consistió en pruebas paramétricas (prueba t de Student) y no paramétricas (Chi-cuadrado y Mann-Whitney), con una p<0,05 para los resultados significativos.
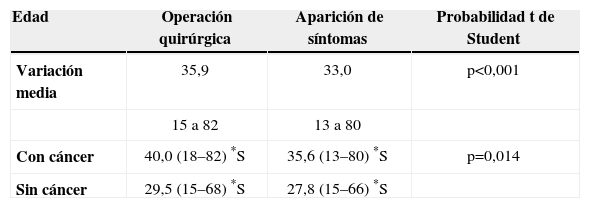
ResultadosEdad, sexo, síntomas e historia familiarSe evaluaron los datos clínicos de 88 pacientes: 41 hombres (46,6%) y 47 mujeres (53,4%). La edad promedio fue de 33,0 años (de 13 a 80) al comienzo de los síntomas y de 33,7 años (de 10 a 80) en el momento del diagnóstico (tabla 1). Durante el tratamiento quirúrgico, la edad media fue de 35,9 años (de 15 a 82), siendo inferior en pacientes sin CCR [29,5 años (de 15 a 68)] comparados con aquellos con CCR [40,0 años (de 18 a 82)] (p<0,001).
Edad de los pacientes en el momento de la operación quirúrgica y de la aparición de síntomas
| Edad | Operación quirúrgica | Aparición de síntomas | Probabilidad t de Student |
| Variación media | 35,9 | 33,0 | p<0,001 |
| 15 a 82 | 13 a 80 | ||
| Con cáncer | 40,0 (18–82) *S | 35,6 (13–80) *S | p=0,014 |
| Sin cáncer | 29,5 (15–68) *S | 27,8 (15–66) *S |
*S: estadísticamente significativo.
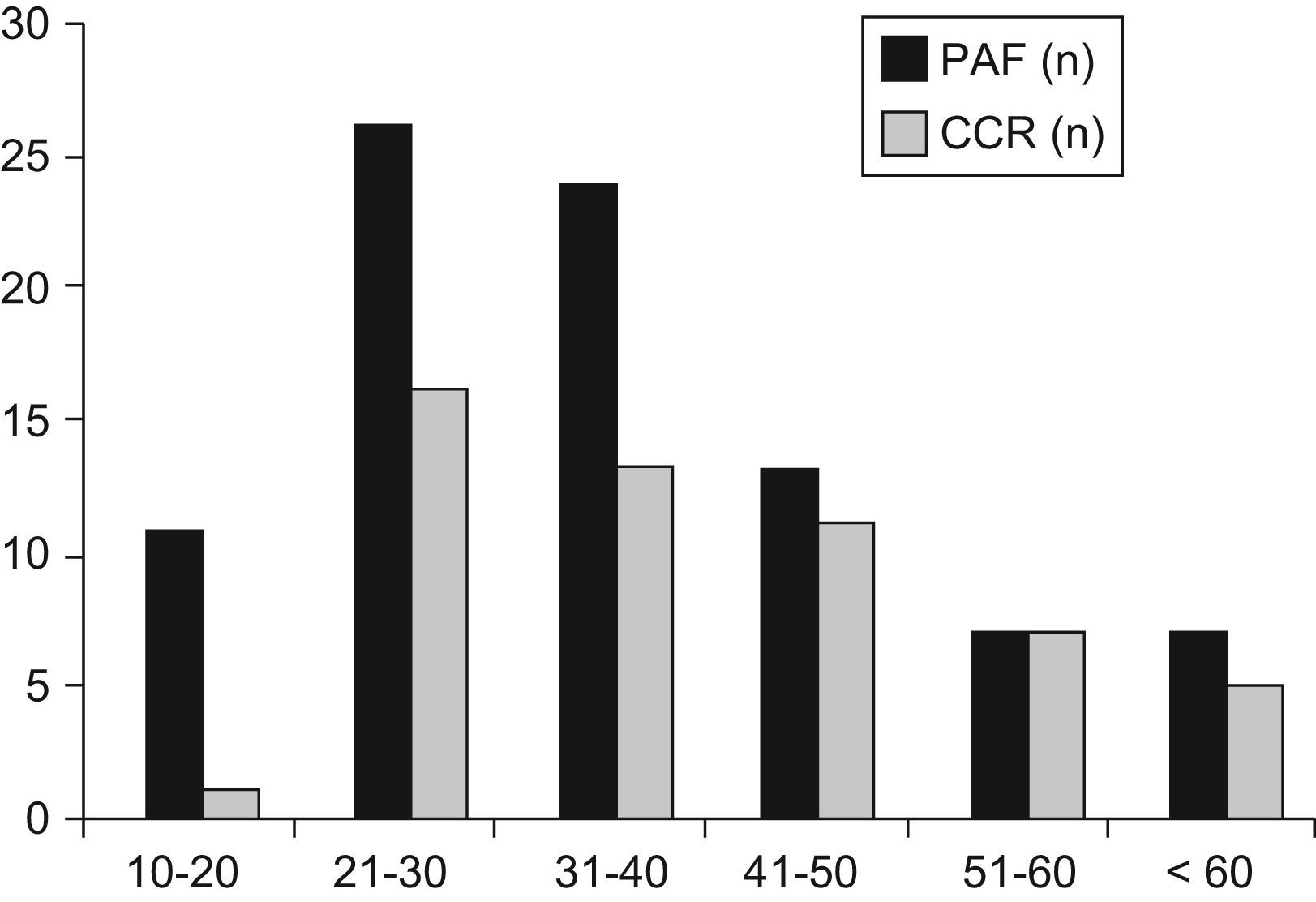
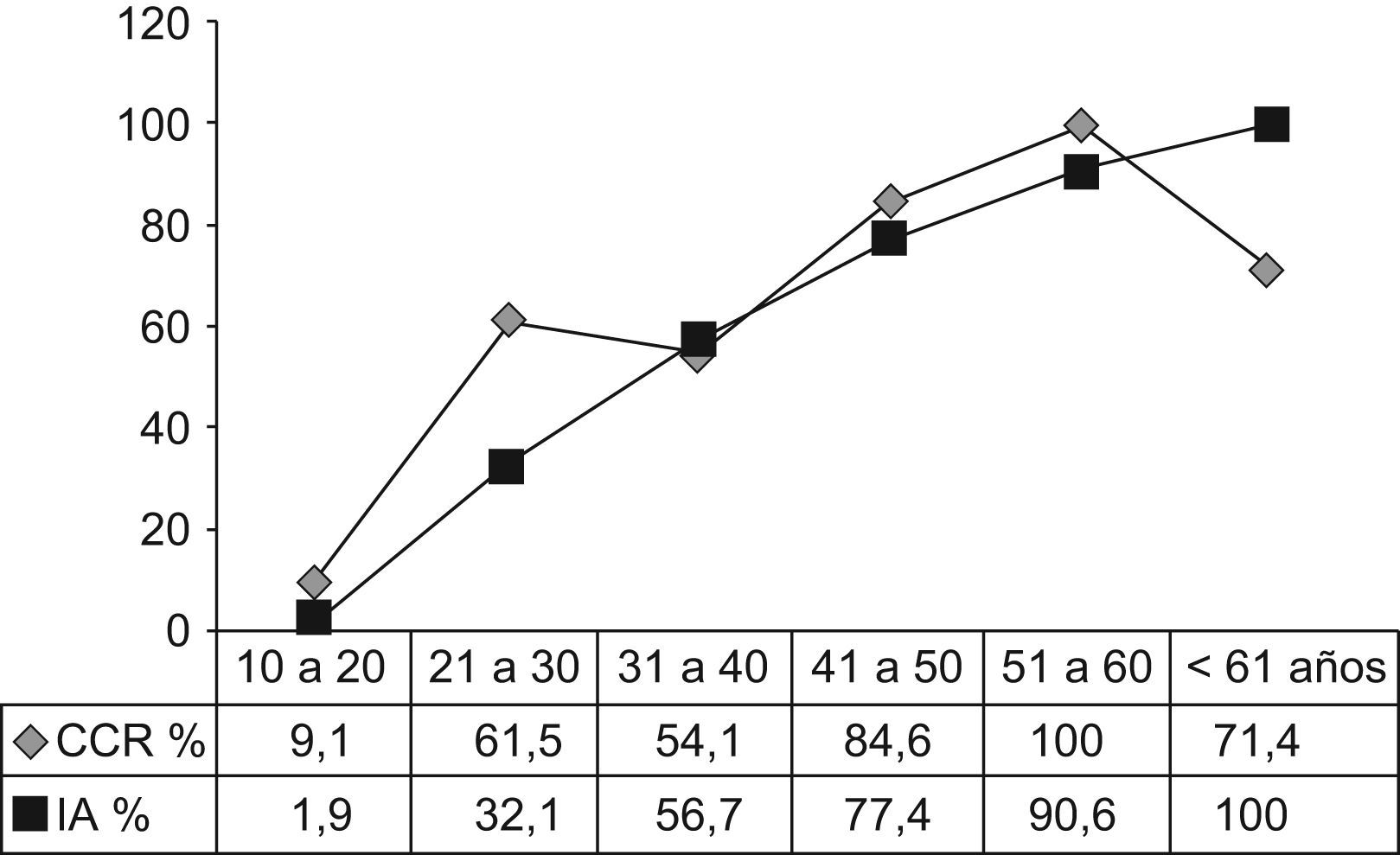
La tabla 2 muestra la distribución de los pacientes con PAF por edad. La incidencia de la enfermedad antes de los 20 años, de los 21 a los 40 años y después de los 40 años fue del 12,5%, 56,8% y 30,7%, respectivamente. Siete pacientes (7,9%) eran mayores de los 60 años. La incidencia de CCR aumentó del 9,1%, antes de los 20 años, al 58% en los pacientes entre 21 a 40 y al 85% después de los 41 años de edad (p<0,001). Las figuras 1 y 2 indican que las incidencias brutas y acumuladas de CCR aumentan de forma significativa después del segundo decenio de la vida.


Unos 58 pacientes (65,9%) notificaron antecedentes familiares de PAF, pero solo 21 (23,8%) fueron diagnosticados debido a familiares afectados. Otros 67 pacientes (76,1%) fueron diagnosticados sobre la base de síntomas relacionados con la poliposis. Los grupos con o sin antecedentes familiares no presentaron ninguna diferencia estadística con respecto a la edad (edad mediana 33,4 años frente a 34,4) ni con la incidencia de CCR (32/58; 55,2% frente a 21/30; 70%) [p=0,17].
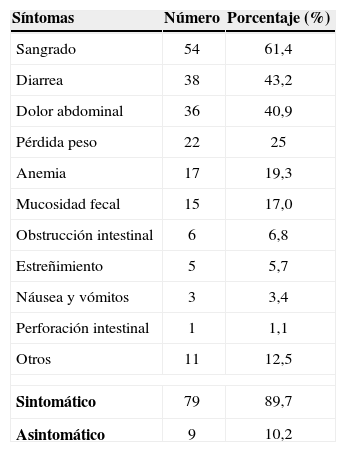
En el momento del diagnóstico, 53 pacientes (60,2%) ya tenían CCR asociado con la poliposis y solo 9 (10,2%) no mostraban síntomas clínicos. Los síntomas más comunes (tabla 3) eran la hemorragia intestinal (61,4%), la diarrea (43,2%), el dolor abdominal (40,9%) y la pérdida de peso (25,0%).
Signos y síntomas clínicos en 79 pacientes sintomáticos
| Síntomas | Número | Porcentaje (%) |
| Sangrado | 54 | 61,4 |
| Diarrea | 38 | 43,2 |
| Dolor abdominal | 36 | 40,9 |
| Pérdida peso | 22 | 25 |
| Anemia | 17 | 19,3 |
| Mucosidad fecal | 15 | 17,0 |
| Obstrucción intestinal | 6 | 6,8 |
| Estreñimiento | 5 | 5,7 |
| Náusea y vómitos | 3 | 3,4 |
| Perforación intestinal | 1 | 1,1 |
| Otros | 11 | 12,5 |
| Sintomático | 79 | 89,7 |
| Asintomático | 9 | 10,2 |
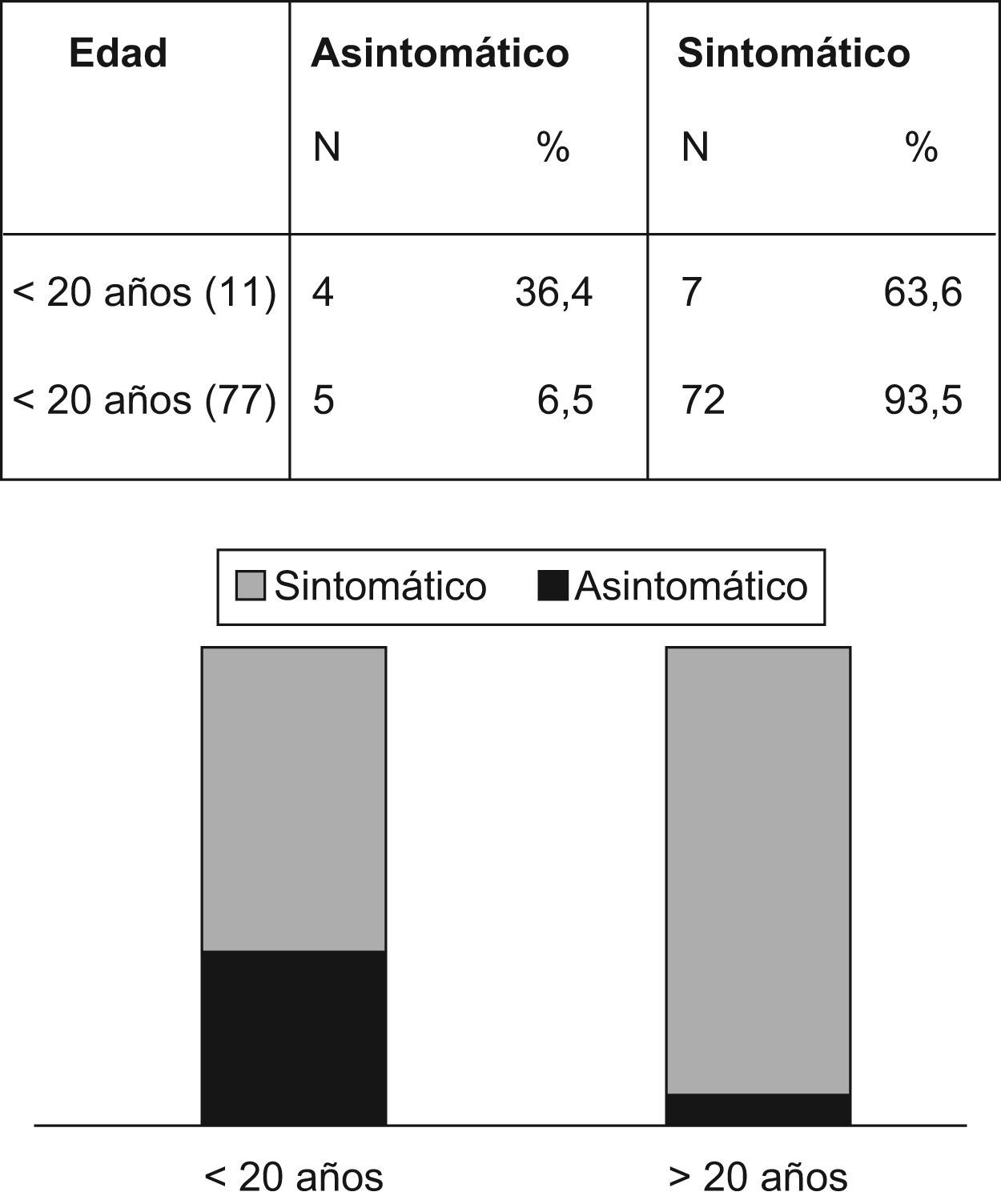
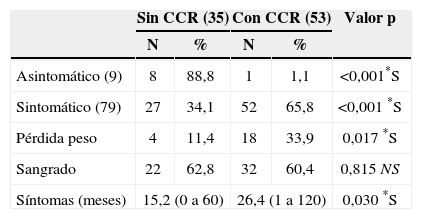
La figura 3 revela que el porcentaje de pacientes asintomáticos se reduce de manera significativa de un 36,4% a un 6,5% después de los 20 años de edad. La tabla 4 indica que significativamente se detectó CCR con menos frecuencia entre los pacientes asintomáticos (n=1; 1,1%) comparados con aquellos que presentaban síntomas (n=52; 65,8%) (p<0,001). La duración promedio de los síntomas era inferior entre los pacientes sin cáncer (15,2 meses frente a 26,4 meses; p=0,03). La pérdida de peso fue un síntoma notificado por 18 pacientes (33,9%) con CCR y por sólo 4 pacientes (11,4%) sin CCR (p=0,017). La hemorragia intestinal estaba presente tanto en pacientes con (n=32/53; 60,4%) como sin CCR (n=22/35; 62,8%) (p=0,8).

Síntomas clínicos en pacientes con y sin cáncer colorrectal (CCR). Los valores se comparan con chi cuadrado y prueba de Mann-Whitney
| Sin CCR (35) | Con CCR (53) | Valor p | |||
| N | % | N | % | ||
| Asintomático (9) | 8 | 88,8 | 1 | 1,1 | <0,001*S |
| Sintomático (79) | 27 | 34,1 | 52 | 65,8 | <0,001 *S |
| Pérdida peso | 4 | 11,4 | 18 | 33,9 | 0,017 *S |
| Sangrado | 22 | 62,8 | 32 | 60,4 | 0,815 NS |
| Síntomas (meses) | 15,2 (0 a 60) | 26,4 (1 a 120) | 0,030 *S | ||
NS: no significativo; *S: estadísticamente significativo.
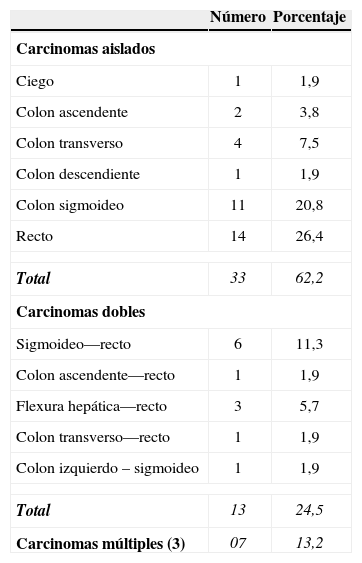
La colonoscopia y la patología en muestras detectaron la presencia de CCR en 53 pacientes (60,2%). Se encontró uno, dos o más de tres tumores en 33 (62,2%), 13 (24,5%) y 7 pacientes (13,2%), respectivamente. Las lesiones aisladas se encontraron principalmente en el recto (14) y en el colon sigmoideo (11) (tabla 5).
Distribución topográfica de 53 carcinomas colorrectales en pacientes con poliposis adenomatosa familiar
| Número | Porcentaje | |
| Carcinomas aislados | ||
| Ciego | 1 | 1,9 |
| Colon ascendente | 2 | 3,8 |
| Colon transverso | 4 | 7,5 |
| Colon descendiente | 1 | 1,9 |
| Colon sigmoideo | 11 | 20,8 |
| Recto | 14 | 26,4 |
| Total | 33 | 62,2 |
| Carcinomas dobles | ||
| Sigmoideo—recto | 6 | 11,3 |
| Colon ascendente—recto | 1 | 1,9 |
| Flexura hepática—recto | 3 | 5,7 |
| Colon transverso—recto | 1 | 1,9 |
| Colon izquierdo – sigmoideo | 1 | 1,9 |
| Total | 13 | 24,5 |
| Carcinomas múltiples (3) | 07 | 13,2 |
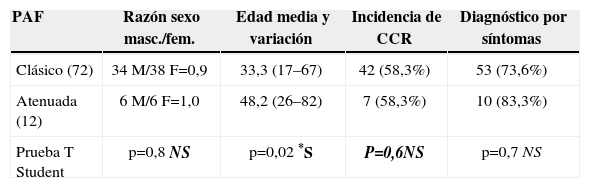
La caracterización de la gravedad de la poliposis mediante colonoscopia fue posible en 84 pacientes. Una pauta atenuada (PAFA) se observó en 12 pacientes (14,3%), el resto se consideró poliposis clásica o grave (n=72; 85,7%). La tabla 6 presenta una comparación de los datos clínicos de PAF y PAFA. Comparados con los pacientes que presentan características clásicas de PAF, la edad promedio en la poliposis atenuada fue superior (48,2 frente a 33,3 años) y la incidencia de CCR similar (58,3% frente a 58,3%). Para la mayoría de los pacientes en ambos grupos (83,3% y 73,6%, p=0,7), el diagnóstico de poliposis se estableció sobre la base de los síntomas. Dos pacientes con fenotipo atenuado desarrollaron cáncer rectal metacrónico tras una anastomosis ileorrectal.
Características clínicas de las formas atenuadas y clásicas de poliposis
| PAF | Razón sexo masc./fem. | Edad media y variación | Incidencia de CCR | Diagnóstico por síntomas |
| Clásico (72) | 34M/38 F=0,9 | 33,3 (17–67) | 42 (58,3%) | 53 (73,6%) |
| Atenuada (12) | 6M/6 F=1,0 | 48,2 (26–82) | 7 (58,3%) | 10 (83,3%) |
| Prueba T Student | p=0,8 NS | p=0,02 *S | P=0,6NS | p=0,7 NS |
CCR: cáncer colorrectal; F: sexo femenino; M: sexo masculino; NS: no significativo; *S: estadísticamente significativo.
La PAF clásica se caracteriza por el desarrollo de cientos o miles de adenomas durante la pubertad, aunque la enfermedad generalmente no se manifiesta hasta el final de la niñez o el principio de la vida adulta. No obstante, se ha observado una variación amplia en los umbrales de edad inicial respecto a las manifestaciones clínicas2,5. La característica clínica más importante es la posibilidad de padecer CCR a una edad temprana. En este contexto, la edad en el momento del diagnóstico se convierte en un parámetro de riesgo predictivo para CCR y debe tenerse en cuenta para elegir el mejor momento para la cirugía.
En nuestra serie, los síntomas empezaron a una edad media de 33 años y el tratamiento quirúrgico se realizó a los 35,9 años; estos datos coinciden con otras medias de edad publicadas: 33 años25 y 34 años26. La distribución de la edad fue del 12,5% antes de los 20 años, del 56,8% entre los 21 y los 40 años y del 30,7% en aquellos con más de 41 años. En el estudio de Croner et al26 se observaron incidencias de un 15%, 53% y 32% para esas mismas edades.
Hoy se admite sin lugar a dudas que la exploración preventiva de la familia es la mejor manera de lograr un diagnóstico y tratamiento tempranos. Este hecho se pone de manifiesto en la baja edad media de los pacientes sin CCR asociado [29,5 años (de 15 a 68 años)] comparado con aquellos con CCR (40 años, de 18 a 82).
Según se muestra en la figura 2, la incidencia bruta de CCR aumentó de un 9,1%, en pacientes con menos de 20 años, a 58% en los de 21 a 40 años y del 85% en individuos de más de 41 años. Resultados similares fueron publicados por Croner et al26 en una serie de 143 pacientes, en la que no había pacientes con CCR por debajo de los 20 años, mientras que las incidencias en los grupos entre 20 y 40 años de edad y los mayores de 40 eran del 30,2% (23/76) y del 65% (30/46), respectivamente.
El análisis de un gran número de familias con PAF desde 1970 a 198027 demostró que el riesgo de CCR antes de los 20 años es muy bajo. Datos similares del Leeds Castle Polyposis Group28 mostraron solo un paciente menor de 15 años. Una revisión reciente29 indicó que la proporción de PAF-pacientes con CCR diagnosticado antes de los 20 años de edad en 1.073 pacientes procedentes de cinco registros europeos era del 1,6%. En nuestras series (fig. 2), encontramos una incidencia acumulada del 1,9% y del 32,1% en los pacientes con menos de 20 y 30 años respectivamente. En resumen, estos datos sugieren que la indicación quirúrgica para la PAF clásica puede demorarse hasta la edad de los treinta a menos que se detecte alguna lesión sospechosa.
Además, esta información respalda la denominada «anastomosis ileoanal en etapas» (anastomosis ileorrectal y proctectomía con reservorio después de un período de 10–15-años) para planificar la estrategia quirúrgica y el momento idóneo para someter a cirugía a los pacientes jóvenes con PAF. La justificación de este enfoque se basa en el hecho de que algunos pacientes pueden evitar complicaciones funcionales, reproductoras y de estomas asociadas con la proctocolectomía restauradora30,31, junto con una reducción importante de patologías desmoides en los pacientes con riesgo32.
La incidencia de CCR en los pacientes diagnosticados fuera de los programas de detección precoz puede ser superior al 60%33. Nosotros diagnosticamos CCR asociado en un 60,2% de nuestros pacientes, ya que la mayoría de ellos fueron diagnosticados sobre la base de síntomas clínicos en lugar de detección precoz. En la bibliografía especializada, la mayoría de los estudios muestran una mayor incidencia de CCR (del 50% al 70%) en los pacientes sintomáticos que en aquellos que llegaron por programas de detección precoz (del 3% al 10%)7,27,34–36.
Diagnosticamos PAF sobre la base de historia familiar en solo el 24% de los pacientes, y el diagnóstico clínico se estableció mediante los síntomas en la gran mayoría (76%): estos datos coinciden con los de Croner et al26 que detectaron PAF por los síntomas en el 84% de sus pacientes. Ninguno de los grupos presentaba diferencias estadísticas con respecto a la edad o la incidencia de CCR, aunque había una tendencia pequeña hacia una mayor proporción de CCR entre aquellos sin historia familiar (70% frente al 55%, p=0,1).
Aunque la historia familiar condujo al diagnóstico del síndrome en casi una cuarta parte de los pacientes, este hecho no influyó en su decisión de introducir un programa de detección precoz que pudiera ayudar a establecer un diagnóstico temprano. En consecuencia, la media de la edad en ambos grupos no fue significativamente diferente en el momento de diagnóstico, y esta característica probablemente tuvo una repercusión mayor sobre la incidencia de CCR a pesar de ser conscientes de la historia familiar. Si una mayor proporción de pacientes con antecedentes familiares positivos hubiera participado en pruebas activas de detección precoz a una edad adecuada (en la mayoría, durante la segunda década de su vida), hubiera sido posible apreciar la influencia de la información familiar que conduce a una asociación reducida con CCR.
A pesar de la presencia de pólipos, los síntomas clínicos pueden ser difusos o incluso ausentes durante los estadios iniciales de la enfermedad. Por este motivo, la evaluación genética o la detección endoscópica debe iniciarse en la pubertad, antes de que los síntomas y el cáncer colorrectal ocurran. La rectorragia es generalmente la primera manifestación, que se vuelve más frecuente e intensa con el transcurso del tiempo. A medida que progresa la enfermedad, los pólipos pueden aumentar en número y tamaño, y los pacientes pueden presentar diarrea, mucosidad, dolor abdominal, anemia, pérdida de peso y estreñimiento. Sin embargo, es posible que estos síntomas no aparezcan hasta que la condición se haya vuelto cancerosa. La diarrea, sangre y mucosidad se consideran una alerta de aparición del cáncer9.
En nuestra serie, la duración media de los síntomas fue más corta (15,2 frente a 26,4 meses) en los pacientes sin cáncer, comparados con aquellos con cáncer. Además, encontramos una incidencia significativa más baja de cáncer entre los pacientes asintomáticos (1,1% frente al 65,8%). De igual manera, Bülow37 informó sobre la existencia de CCR en un 67% de los probandos (diagnosticados sobre la base de síntomas, sin conocer la enfermedad hereditaria) contra un 3% detectado mediante técnicas de detección precoz en familias en Dinamarca. En otros países, los datos de los registros nacionales de poliposis mostraron resultados similares en los Países Bajos (47% frente al 4%)36, Finlandia (65,5% frente al 6,6%)38, Australia (75% frente al 6%)39 e Italia (80% frente al 3,3%)40. Estos números demuestran claramente la utilidad de la detección precoz en la incidencia de CCR asociada con el síndrome.
Además de la edad, la presencia de síntomas clínicos, la pérdida de peso y la duración de síntomas se asociaron también con un riesgo mayor de CCR. Observamos que 18 de 53 pacientes (33,9%) con CCR informaron sobre la pérdida de peso, mientras que solo 4 individuos (11,4%) sin CCR lo hicieron (p=0,01). Por otro lado, la hemorragia intestinal fue notificada con igual frecuencia por los pacientes con CCR (n=32/53; 60,4%) y sin CCR (n=22/35; 62,8%).
Treinta y tres de nuestros pacientes presentaban solo un tumor (62,2%), principalmente ubicado en el recto (14) y el sigma (11). Debe señalarse que un número significativo de pacientes tenía dos (24,5%) o más tumores (13,2%). Este hecho tiene importantes implicaciones terapéuticas y pronósticas, ya que plantea la necesidad de la resección oncológica de todos los segmentos durante la colectomía profiláctica. Otra serie26 observó un predominio del cáncer de colon (59,5%) sobre el cáncer rectal (40,5%), similar a nuestros resultados en pacientes con lesiones en la muestra (57,6% y 42,4%, respectivamente).
La posibilidad de indicar un tratamiento quirúrgico en la ausencia de CCR depende de diversos factores como la edad, las características genómicas y también la gravedad de la poliposis. Se admite que la presencia de más de 1.000 pólipos duplica el riesgo de CCR41, y estos fenómenos están relacionados con el sitio exacto de la mutación donde algunas regiones (hotspots) como el codon 1309 conducen a enfermedad grave42–47 y comienzo temprano de los síntomas10,27,28,34. Cuando no se trata, la muerte de los pacientes con CCR y una mutación en el codon 1309 ocurre unos 10 años antes de la media34.
La poliposis adenomatosa familiar atenuada (PAFA), anteriormente denominada síndrome hereditario de adenoma plano48, está causada por mutaciones específicas y su frecuencia no está aún definida. Estos pacientes desarrollan CCR unos 10 o 15 años después de aquellos con PAF clásica12, pero antes que los pacientes con CCR esporádico49,50.
Entre nuestros pacientes, se obtuvo la caracterización de la gravedad de la poliposis mediante colonoscopia en 84 pacientes, de los que 12 (14,3%) fueron definidas como atenuada y 72 (85,7%) como clásica. La comparación de los dos grupos reveló que la edad media era mayor entre los pacientes con PAFA (48,2 años frente a 33,3 años) y la incidencia de CCR asociado similar (58,3% frente al 58,3%). Es importante observar que el diagnóstico se estableció en ambos grupos sobre la base de los síntomas en lugar de los antecedentes familiares (83,3% y 73,6%, respectivamente; p=0,7). Por lo tanto, este resultado resalta nuevamente el valor de la duración de los síntomas aun en pacientes con una enfermedad más leve.
Durante el seguimiento, fue posible detectar cáncer rectal en seis pacientes (16,6%) después de anastomosis ileorrectal. Como dos de ellos habían sido diagnosticados anteriormente con la variedad atenuada a los 47 y 56 años de edad, es evidente que los pacientes con PAFA tampoco están libres del riesgo de cáncer rectal metacrónico, principalmente cuando se relaciona con una edad avanzada51.
Todos los datos aportados despiertan la atención sobre la mayor incidencia de CCR en los pacientes diagnosticados por una vía distinta a las pruebas de detección precoz. También sugieren que la edad es un factor indicativo crucial para el riesgo de CCR, por lo que se convierte en una guía natural para definir el mejor momento para el tratamiento quirúrgico. Nuestros resultados indican que el riesgo de CCR en pacientes con PAF aumenta de forma significativa después de los veinte años de vida, y se asocia con otras características clínicas tales como la pérdida de peso y la duración de los síntomas.
La PAF es una enfermedad rara y compleja que presenta múltiples expresiones clínicas. Por lo tanto, un único enfoque de manejo no servirá para atender las exigencias de todos los pacientes, lo que convierte el tratamiento de la PAF un tema para especialistas. La decisión de cómo y cuándo operar puede resultar especialmente difícil con pacientes jóvenes que deben ser convencidos para que se sometan a una colectomía profiláctica que con el tiempo podría alterar su imagen corporal, la fecundidad y las funciones de evacuación.
Teniendo en cuenta este escenario tan difícil, una exposición adecuada de todos los problemas por parte del especialista puede facilitar la comprensión de los riesgos y los detalles necesarios para tomar una decisión quirúrgica. La opción del procedimiento quirúrgico debe basarse principalmente en la gravedad de la enfermedad. Por lo tanto, la mayoría de los pacientes con pocos adenomas rectales, un fenotipo colónico leve, ningún carcinoma colorrectal, antecedentes familiares de fenotipo leve y aquellos con formas atenuadas (poliposis asociada con PAFA y MAPA-MUTYH ) responden bien con una anastomosis ileorrectal, pero los pacientes con poliposis grave necesitarán una proctocolectomía restauradora20,51.
En segundo lugar, la decisión debe ser personalizada y tener en cuenta la edad, el estado de salud y las preferencias personales33,52. Aun en los casos con poliposis grave, la anastomosis ileorrectal es una buena opción para mujeres jóvenes que deseen quedarse embarazadas en el futuro, aunque debe recalcarse la necesidad de mantener una estrecha vigilancia y el riesgo de finalmente tener que ser sometidas a una proctectomía con anastomosis ileoanal52.
Cuando exista, la información genética puede ayudar a tomar una decisión final. Una investigación reciente ha mostrado que un análisis de las mutaciones puede predecir la necesidad de proctectomía secundaria53. No obstante, la evaluación de los datos clínicos y endoscópicos deben ayudar al cirujano a decidir cuándo y cómo operar a los pacientes dependiendo de sus características.
Así, el riesgo de CCR en los pacientes con PAF 1) aumenta de forma significativa después de los 20 años de edad; 2) se asocia con una edad mayor así como una pérdida de peso, presencia y duración de la sintomatología superiores; 3) es similar en los pacientes con fenotipo atenuado y clásico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Parte de este trabajo se presentó en el Congreso Brasileño de Coloproctología de 2009 (7–9 de septiembre en São Paulo, Brasil).

