






Introducción. En los últimos años, múltiples estudios epidemiológicos y experimentales han demostrado que la ingesta de una dieta rica en fibra disminuye la incidencia y mortalidad por cáncer colorrectal. Estudios in vitro han demostrado que el ácido butírico, un ácido graso de cadena corta derivado de la fermentación de la fibra dietética en el colon, bloquea la proliferación de células tumorales derivadas de cáncer de colon, por lo que se cree que es el principal componente protector de las dietas ricas en fibra en la carcinogénesis colónica.
Métodos. En este artículo hemos realizado una revisión exhaustiva de la bibliografía. Tras un breve resumen del modelo genético del cáncer colorrectal, analizamos las propiedades antitumorales del butirato, así como el estado actual de conocimiento de su mecanismo de acción molecular.
Resultados. La carcinogénesis es un proceso multietapa caracterizado por la expansión del área de proliferación, la alteración del patrón de diferenciación y la disminución de la apoptosis. En cultivos celulares derivados de cáncer de colon se ha observado que el butirato, a concentraciones fisiológicas, inhibe la proliferación celular bloqueando la célula en fase G1 del ciclo celular, induce diferenciación y apoptosis, y modula la expresión de múltiples genes, incluidos algunos de los oncogenes y genes supresores implicados en la carcinogénesis colorrectal.
Conclusión. Actualmente se cree que el butirato es el principal componente protector de la fibra dietética en la carcinogénesis colorrectal, al inducir detención de crecimiento, diferenciación y apoptosis. Sin embargo, se necesita realizar más estudios para determinar el mecanismo exacto de acción molecular.
Introduction. In the last few years, many epidemiological and experimental studies have demonstrated that ingestion of a high-fiber diet diminishes the incidence and mortality of colorectal cancer. In vitro studies have shown that butyric acid, a short chain fatty acid derived from the fermentation of dietary fiber in the colon, blocks the proliferation of colon cancer tumor cells. Consequently, butyric acid is believed to be the main protective component against colonic carcinogenesis in high-fiber diets.
Methods. This article provides an exhaustive review of the literature. After brief summarizing the genetic model of colorectal cancer we analyze the anti-tumor properties of butyrate as well as the current state of knowledge of its molecular mechanism of action.
Results. Carcinogenesis is a multi-stage process characterized by the expansion of the proliferation area, alterations in pattern of differentiation and diminution of apoptosis. In colon cancer cell cultures, physiological concentrations of butyrate inhibit cellular proliferation, blocking the cell at stage G1 of the cell cycle, induce differentiation and apoptosis and modulate the expression of multiple genes, including some of the oncogenes and tumor suppressor genes involved in colorectal carcinogenesis.
Conclusion. Butyrate is currently believed to be the main protective component in dietary fiber against colorectal cancer, reducing growth, differentiation and apoptosis. However, further studies are needed to determine its precise molecular mechanism of action.
Introducción
Después de aproximadamente dos décadas de estudios moleculares, existen pocas dudas de que el cáncer es una enfermedad genética, y se acepta que la causa fundamental del mismo reside en alteraciones de genes específicos relacionados con aspectos críticos de la función celular (proliferación y muerte celular)1. Por lo general, las mutaciones en estos genes se acumulan en células somáticas durante años hasta que una célula pierde un número crítico de mecanismos de control del crecimiento e inicia la formación de un tumor. La frecuencia de estas mutaciones puede ser modificada por la exposición a factores ambientales, lo que establece un vínculo entre estos últimos y los factores genéticos.
Así pues, ni la genética ni el entorno son determinantes únicos de la carcinogénesis. Aunque aún no conocemos la causa precisa que origina el crecimiento tumoral, sí podemos afirmar que posiblemente el carcinoma colorrectal tenga un origen multifactorial que incluye factores medioambientales (sobre todo dietéticos) y alteraciones genéticas, tanto heredadas como somáticas2.
Alteraciones genéticas. Modelo genético del carcinoma colorrectal
Desde la identificación del locus APC en pacientes con poliposis adenomatosa familiar hasta los más recientes descubrimientos de los genes implicados en la reparación del ADN, se ha recorrido un largo camino en el que se han ido identificado genes implicados en las diferentes etapas del desarrollo del carcinoma colorrectal. En su conjunto se encuentran representados los tres tipos de genes tumorales, oncogenes, genes supresores y genes reparadores de errores de replicación del ADN, implicados en la regulación del ciclo celular, replicación y reparación del ADN, cuyas mutaciones hacen más susceptibles a las células para que se transformen en neoplásicas2.
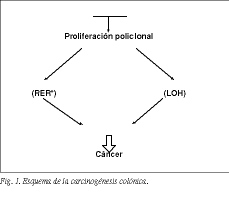
La biología molecular del cáncer colorrectal parece seguir el modelo general de carcinogénesis propuesto por Loeb3, según el cual factores tanto ambientales como del huésped pueden producir la pérdida de los mecanismos de reparación del ADN, lo cual conduce a una inestabilidad genómica que facilita la rápida acumulación de mutaciones somáticas o la pérdida de heterozigosidad dentro de oncogenes y genes supresores tumorales. La activación de oncogenes lleva a la célula a un proceso de expansión clonal, mientras que la inactivación de genes supresores tumorales libera a las células de las limitaciones del crecimiento normal (fig. 1).

Tanto los pólipos como los cánceres son clonales en su naturaleza, habiéndose originado de una única célula madre transformada dentro del compartimiento proliferativo de la cripta colónica. Previo a este evento monoclonal aislado, el colon entero sufre una alteración generalizada (policlonal) o "efecto difuso" caracterizado por una proliferación celular acelerada, así como una expansión del área de replicación celular dentro de las criptas4. En este ambiente, deleciones, mutaciones u otras alteraciones del ADN se acumulan rápidamente en genes específicos relacionados con la tumorogénesis1.
Los cambios genéticos implicados en el proceso neoplásico, la secuencia histológica y los eventos moleculares son similares, tanto si el cáncer se desarrolla en el contexto de un síndrome hereditario, un agrupamiento familiar o esporádico, y se han identificado dos vías moleculares que juntas explican más del 95% de los cánceres colorrectales: progresión tumoral vía error de replicación (RER), en la que el cáncer se inicia por una mutación hereditaria o somática en uno de los genes de reparación del ADN, los denominados genes de reparación del apareamiento incorrecto de bases (MMR)1,3 y progresión tumoral vía pérdida de heterozigocidad (LOH), la cual generalmente se inicia por pérdida alélica y/o mutación puntual del gen APC5.
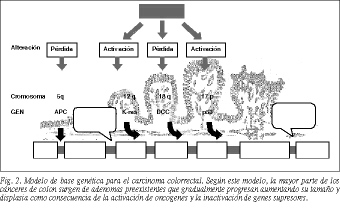
En 1988, Fearon y Vogelstein6, apoyándose en estudios epidemiológicos, histopatológicos, clínicos y de biología molecular, propusieron un modelo de la base genética del cáncer co lorrectal según el cual la mayor parte de los cánceres de colon surgen de adenomas preexistentes, que gradualmente progresan incrementando su tamaño y displasia como resultado de la activación de oncogenes y de la inactivación de genes supresores (fig. 2).

Las alteraciones genéticas más precoces que se han identificado en el cáncer colorrectal ocurren generalmente dentro del gen APC5,6 y/o en uno de los genes de reparación de los errores de replicación7. Las mutaciones en el APC producen un incremento en la tasa de proliferación de las células madre en la cripta colónica, dotando a cada una de ellas de un potencial aumentado para el crecimiento clonal que puede dar origen a un pequeño adenoma4. Los eventos genéticos responsables de la transición de hiperplasia a neoplasia no están claros8, pero parecen incluir mutaciones en todos estos genes siguiendo en general el orden que se observa en la figura 2. Se cree que con la alteración de K-ras comienza la expansión clonal de un pequeño adenoma2,6,9. Dicho gen codifica una proteína denominada p21 que participa en la transmisión de señales desde los receptores tirosinacinasa en la superficie de la célula hasta el núcleo, estimulando la proliferación y diferenciación celular10. La mutación de K-ras produce una proteína alterada que estimula continuamente la división celular4. Para que un pólipo progrese más allá de este estadio, una única célula dentro del adenoma debe perder la función de un gen supresor tumoral, siendo generalmente el gen DCC en el cromosoma 18q el próximo en ser desactivado o perdido6,9.
Hay pocas dudas acerca de la importancia de las mutaciones del gen p53 en la progresión de los tumores colorrectales, y se han descrito alteraciones del mismo en más del 75% de los cánceres colorrectales11. Sin embargo, dichas mutaciones son infrecuentes en adenomas en cualquier estadio6,12, lo que sugiere que las primeras etapas del desarrollo del cáncer no dependen de la represión de la actividad de p53, la cual estaría implicada en la transición de adenoma a carcinoma6,13. Además, estas mutaciones a menudo se asocian con tumores más agresivos, incluyendo mayor progresión a las metástasis y supervivencias más cortas.
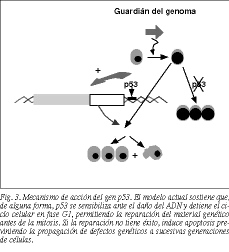
El gen p53 codifica una fosfoproteína nuclear de 53 kD, un factor de transcripción nuclear implicado en la síntesis y reparación del ADN, el control del ciclo celular (es un regulador negativo de la proliferación celular), la diferenciación celular y la muerte celular programada o apoptosis14,15. Aunque no se conoce de forma precisa su mecanismo de acción, quizás el aspecto más destacado sea el de guardián del genoma16, al desempeñar un importante papel en la reparación del ADN. El modelo actual sostiene que, de alguna forma, p53 se sensibiliza ante el daño del ADN y detiene el ciclo celular en fase G1, y posiblemente también en fase G2, permitiendo que la reparación del material genético tenga lugar antes del comienzo de la síntesis de ADN y la mitosis, disminuyendo por lo tanto la probabilidad de mutaciones17-19. Si la reparación no tiene éxito o el daño es muy severo, puede promover la muerte celular por apoptosis, previniendo de esta forma la propagación de defectos genéticos a sucesivas generaciones de células18 (fig. 3).

Por otra parte, p53 induce la transcripción de varios genes implicados en el control del ciclo celular. Entre estos destaca el gen p21/WAF1, el cual codifica un potente inhibidor de las cinasas dependientes de ciclinas responsable de la inactivación de la proteína Rb, un paso fundamental para la detención de las células en las fases G1 y G2 y la apoptosis17,19-22.
Las mutaciones de p53 producen unas proteínas alteradas, la mayoría de las cuales son incapaces de unirse al ADN dañado, con lo que las células no se detienen en fase G1 y prosiguen el ciclo celular sin haber reparado las lesiones genómicas23 (fig. 3).
Factores ambientales
Desde los trabajos de Burkitt24, múltiples estudios epidemiológicos y en animales de experimentación han demostrado que la ingesta de una dieta rica en fibra y compuestos hidrocarbonados se relaciona con un menor riesgo y mortalidad por cáncer co lorrectal25,26. La fibra, el almidón resistente y los fructosoligosacáridos escapan a la digestión en el estómago e intestino delgado y sufren un proceso de fermentación anaerobia en el colon, produciendo ácidos grasos de cadena corta (AGCC), fundamentalmente ácido acético, ácido propiónico y ácido butírico27,28. Aproximadamente el 95-99% de los AGCC son rápidamente absorbidos y metabolizados por los colonocitos, para los que son su principal fuente de energía, ejerciendo además otras acciones entre las que se incluyen aumento del flujo sanguíneo colónico, aumento de la absorción de sodio y agua, estimulación de la producción de moco y disminución del pH en la luz del colon28,29.
De los tres se ha observado que es el ácido butírico el que tiene los efectos más importantes sobre las células epiteliales colónicas, tanto in vitro como in vivo, por lo que actualmente se le reconoce como el principal componente protector de las dietas ricas en fibra frente a la carcinogénesis colónica25,28,30.
Propiedades antitumorales del butirato
El principal papel del butirato en relación con su propiedad antitumoral radica en sus efectos sobre el crecimiento, diferenciación y apoptosis de los colonocitos. En este sentido, ejerce una serie de acciones sobre las que se fundamenta dicho efecto antineoplásico:
1. Detiene el crecimiento de los colonocitos neoplásicos en una gran variedad de líneas celulares derivadas de cáncer de colon.
El epitelio colónico es un sistema dinámico con células madre localizadas en la base de la cripta colónica que proliferan y migran hacia la superficie de la misma, donde se diferencian completamente, siguiendo en último lugar muerte celular por apoptosis. El proceso de la carcinogénesis rompe esta migración ordenada, de manera que las células continúan la proli feración y migración hacia la superficie de las criptas, no presentando las características de diferenciación normal y, ocasionalmente, invadiendo la membrana basal31.
Aunque los mecanismos que regulan esta proliferación, diferenciación y apoptosis están intrínsecamente programados, también están sujetos a la modulación por factores externos como los AGCC. El butirato, a concentraciones fisiológicas de 1-10 mmol/l, detiene el crecimiento de los colonocitos neoplásicos en fase G1 del ciclo celular en múltiples líneas celulares en cultivo32-34 e in vivo, donde la experiencia es más limitada35.
2. Induce la expresión de marcadores de diferenciación tanto in vitro como in vivo34. Así, aumenta la actividad de enzimas del borde en cepillo de la membrana plasmática, como la fosfatasa alcalina y la dipeptidil peptidasa IV36,37 (marcadores de diferenciación cuya expresión disminuye con la transformación neoplásica), la expresión del antígeno cluster-1 del cáncer de pulmón de células pequeñas, asociado con el aumento de la diferenciación de algunas líneas celulares de adenocarcinoma, y del CEA38.
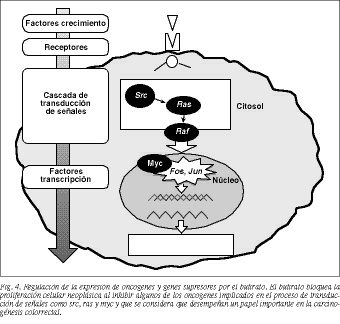
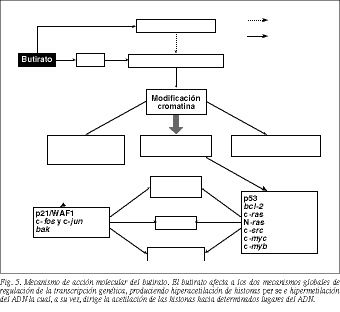
3. Actualmente se cree que la carcinogénesis colorrectal es un proceso multietapa que resulta, en parte, de la acumulación de alteraciones en determinados genes, oncogenes y genes supresores, cuyas proteína-productos forman parte de la red de transmisión de señales que operan desde la membrana citoplasmática hasta el núcleo y que controlan la proliferación y diferenciación celular (tabla 1)39. El butirato bloquea la proliferación celular neoplásica al modificar la expresión de algunos de estos genes implicados en el proceso de transmisión de señales: inhibe los protooncogenes ras40, src41 y c-myc42 e induce la expresión de los protooncogenes c-fos y c-jun4 3 (fig. 4).

4. Induce apoptosis, un acontecimiento importante en la carcinogénesis colorrectal, en varias líneas celulares derivadas de adenomas y carcinomas de colon36,44,45. Esta apoptosis es independiente de p5344-46 y existen evidencias de que ocurre como etapa final del proceso de diferenciación (la apoptosis p53 dependiente en respuesta al daño del ADN ocurre por otra vía distinta de la de la diferenciación terminal)36. En cuanto a su relación con la expresión de proteínas de la familia Bcl-2, ésta es compleja y varía según la línea celular estudiada46. El butirato aumenta las concentraciones de Bak en algunas líneas celulares, mientras que en otras disminuye los valores de Bcl-2, lo cual produce una disminución de la relación Bcl-2/Bax que puede estar implicada en la promoción de la apoptosis46.

Mecanismo de acción del butirato
Aunque el mecanismo exacto responsable de todas estas acciones es en gran parte desconocido, se sabe que el butirato puede actuar a tres niveles:
1. Como cofactor de proteínas nucleares implicadas en la regulación de la transcripción genética47.
2. Interaccionando con proteínas G, componentes clave de la vía de transducción de señales, en la interfase membranocitoplasmática48.
3. Modulando la actividad de varias enzimas nucleares, las cuales a su vez influyen sobre los distintos componentes estructurales de la cromatina, por lo que en último término también modifican la expresión de determinados genes implicados en la proliferación, diferenciación y apoptosis celular. Así, el butirato produce: a) hiperacetilación de histonas por inhibición de la histona desacetilasa (HDAC)49,50; b) inhibición de la fosforilación de las histonas H1 y H247,49-52; c) aumento de la fosforilación de la histona H352; d) hipermetilación de los residuos de citosina del ADN47,49,51,53, y e) aumento de la acetilación y fosforilación de proteínas no histonas y aumento de la ADP ribosilación47,51.
De entre todas estas acciones, la acetilación de las histonas es la más afectada por el butirato, y probablemente es la responsable de la mayoría de sus efectos moleculares. La hiper acetilación reduce el número de cargas positivas susceptibles de unirse a las negativas del ácido fosfórico, con lo que disminuye la afinidad de las histonas por el ADN. La unión entre éstos es más laxa, lo cual produce un cambio conformacional sutil en la estructura del nucleosoma que "abre" la configuración del ADN54, lo que conlleva una serie de consecuencias (fig. 5):

Aumento de la accesibilidad a la cromatina de las ADNasas54,55, las cuales rompen el ADN por unos lugares específicos o puntos hipersensitivos la mayoría de los cuales están relacionados con la expresión génica. Por otro lado, este aumento de la digestión por las ADNasas puede ser responsable, en parte, de la fragmentación de los nucleosomas, un paso clave y precoz en el proceso apoptósico56.
Favorece la entrada de los factores encargados de ejecutar los procesos de reparación del ADN57.
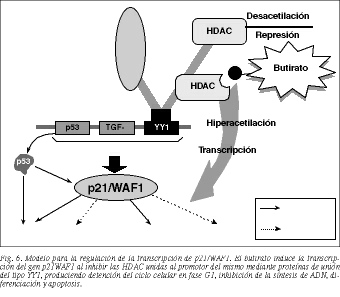
El principal efecto molecular del butirato parece radicar en la inducción de la transcripción de determinados genes al facilitar la llegada de ciertos factores de transcripción a sus lugares de unión en el ADN53,58. Los inhibidores de la HDAC estimulan, entre otros, la transcripción del gen p21/WAF153,58,59, por lo que los efectos del butirato sobre la célula están estrechamente relacionados con la expresión de este gen60,61. Tanto la inhi bición de la HDAC como la expresión de p21 producen de tención del ciclo celular en fase G1, diferenciación y apoptosis53,59-61, lo que sugiere que la inducción de p21/WAF1 es un evento central para estas acciones llevadas a cabo por el buti rato.
El promotor del gen p21/WAF1 posee varios lugares de unión para distintos factores que activan su transcripción: p53, en respuesta al daño del ADN17,60, TGFß, suero y otros inductores fisiológicos y químicos de diferenciación, incluido el butirato, en este último caso independientemente de p5359,62. Experimentos recientes han demostrado una unión física entre los inhibidores de la HDAC y otros factores reguladores de la transcripción que, o bien se unen directamente al ADN, o bien están asociados con proteínas de unión al mismo como YY1 (Yin Yang 1)63. El análisis del promotor del gen p21/WAF1 ha revelado que éste presenta un lugar de unión para YY1, el cual se cree que inhibe la transcripción de p21 en parte por reclutamiento de las HDAC en el promotor del gen64. El butirato se une e inhibe a la HDAC permitiendo la transcripción de p21/WAF1 (fig. 6).

Como sabemos, todas las decisiones de proliferación celular son ejecutadas por el sistema de control del ciclo celular, un dispositivo bioquímico basado en dos familias de proteínas: las cinasas dependientes de ciclinas (Cdk) y sus subunidades reguladoras, las ciclinas65. Éstas se unen y forman complejos ciclina/Cdk, los cuales fosforilan determinadas proteínas diana que ejecutan pasos clave en la progresión del ciclo celular66. En la mayoría de las células, el objetivo molecular de estos complejos ciclina/Cdk es la proteína del retinoblastoma pRb66,67, una fosfoproteína nuclear que es un regulador negativo crítico de la proliferación celular. En este sentido, pRb actúa como "guardián" del punto de control G1 en función de su estado de fosforilación, de modo que, en forma hipofosforilada (activa), se une e inhibe a un conjunto de proteínas reguladoras que favorecen la transcripción de determinados genes necesarios para la proliferación celular, como myc y fos, retrasando el avance del ciclo celular, mientras que cuando se fosforila (inactiva) pierde su capacidad de suprimir el crecimiento y el ciclo celular continúa su progresión23.
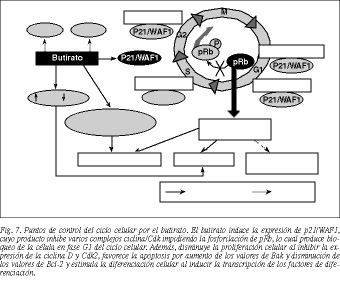
Además de por los complejos ciclina/Cdk, el sistema de control del ciclo celular está regulado por una familia de proteínas inhibidoras de las Cdks llamadas CdkIs20,67, las cuales se unen a los complejos ciclina/Cdk y bloquean su actividad catalítica60. De esta forma no se produce la fosforilación de pRb y la célula se detiene en fase G1, por lo que estas proteínas inhibidoras potencialmente actúan como supresores tumorales17,19,20,68. Dentro de las CdkIs, en relación con el cáncer de colon, es especialmente importante la proteína p21/WAF1, la cual se une e inhibe a una gran cantidad de complejos ci clina/Cdk (ciclina D/Cdk4/6, ciclina E/Cdk2, ciclina B/Cdc2 y ciclinaA/Cdk2)17,20,68, así como a varios componentes de la maquinaria de replicación del ADN, fundamentalmente PCNA22,60, deteniendo a la célula en fase G1 del ciclo celular.
La fosforilación de pRb, iniciada por los complejos ciclina D/Cdk4/6 y completada por las ciclinas E/Cdk2, es crítica para la proliferación alterada de las células malignas. El butirato, al inducir la expresión de p21/WAF1, impide la fosforilación de pRb, con lo que se produce el bloqueo de la célula en fase G1, lo que a su vez conlleva una disminución de la proliferación celular y una inducción de la diferenciación y la apoptosis. Además, el butirato produce otros efectos que contribuyen a la inhibición de la proliferación: disminución de las concentraciones de ciclina D y Cdk269, aumento de los valores de Bak y disminución de los valores de Bcl-246, y estimula la transcripción de factores de diferenciación34 (fig. 7).

Cuál es el papel exacto de cada uno de estos mecanismos y en qué grado se complementan o potencian entre sí es algo que debemos investigar. Por ahora, nuestro grupo de trabajo ha demostrado una importante reducción en la aparición y desarrollo de carcinomas colorrectales en un modelo experimental de cáncer de colon en ratas de laboratorio a las que se les induce tumor mediante la administración de dimetilhidracida (DMH), tanto cuando se les administra butirato por vía intracolónica como por vía intravenosa. Creemos que su mecanismo de acción principal reside en sus efectos en la regulación de la expresión de determinados genes, fundamentalmente de p21/ WAF1, aunque este hecho está aún por demostrar

