


La inflamación sistémica se caracteriza por la elevación en los niveles circulantes de citocinas inflamatorias; así como aumento en la infiltración de macrófagos en tejidos periféricos. Este escenario inflamatorio no induce lesión o pérdida de la funcionalidad en el tejido infiltrado, rasgo distintivo de un estado de inflamación sistémica de grado bajo. La inflamación sistémica de grado bajo posee una estrecha relación con el desarrollo de enfermedades cardiometabólicas en el paciente con obesidad, por lo que este estado de alteración inmune también ha recibido el nombre de metainflamación.
ObjetivoEn esta revisión presentamos la evidencia clínica y experimental más reciente en torno al papel de la inflamación del tejido adiposo como detonante de la metainflamación. Además, revisamos a nivel molecular los mecanismos de polarización inflamatoria de macrófagos invasores del tejido adiposo y el endotelio vascular a través de la activación de receptores tipo toll por patrones moleculares asociados a daño metabólico, tales como proteínas glucosiladas y lipoproteínas oxidadas. Por último, revisamos los mecanismos fisiopatogénicos de la inflamación sistémica en el desarrollo de resistencia a la insulina, dislipidemia, aterogénesis, diabetes mellitus tipo 2 e hipertensión en el paciente obeso.
ConclusionesUn entendimiento más detallado de los mecanismos moleculares a través de los cuales la inflamación sistémica de grado bajo promueve el desarrollo de enfermedades cardiometabólicas podría ser útil en el diseño de terapias antiinflamatorias que tengan en cuenta datos clínicos, así como el nivel circulante de citocinas, células inmunes y patrones moleculares asociados a daño metabólico en el paciente.
Systemic inflammation is characterised by high circulating levels of inflammatory cytokines and increased macrophage infiltration in peripheral tissues. Most importantly, this inflammatory state does not involve damage or loss of function of the infiltrated tissue, which is a distinctive feature of the low-grade systemic inflammation. The term “meta-inflammation” has also been used to refer to the low-grade systemic inflammation due to its strong relationship with the development of cardio-metabolic diseases in obesity.
ObjectiveA review is presented on the recent clinical and experimental evidence concerning the role of adipose tissue inflammation as a key mediator of low-grade systemic inflammation. Furthermore, the main molecular mechanisms involved in the inflammatory polarization of macrophages with the ability to infiltrate both the adipose tissue and the vascular endothelium via activation of toll-like receptors by metabolic damage-associated molecular patterns, such as advanced glycation-end products and oxidized lipoproteins, is discussed. Finally, a review is made of the pathogenic mechanisms through which the low-grade systemic inflammation contributes to develop insulin resistance, dyslipidaemia, atherogenesis, type 2 diabetes, and hypertension in obese individuals.
ConclusionsA better understanding of the molecular mechanisms of low-grade systemic inflammation in promoting cardio-metabolic diseases is necessary, in order to further design novel anti-inflammatory therapies that take into consideration clinical data, as well as the circulating levels of cytokines, immune cells, and metabolic damage-associated molecular patterns in each patient.
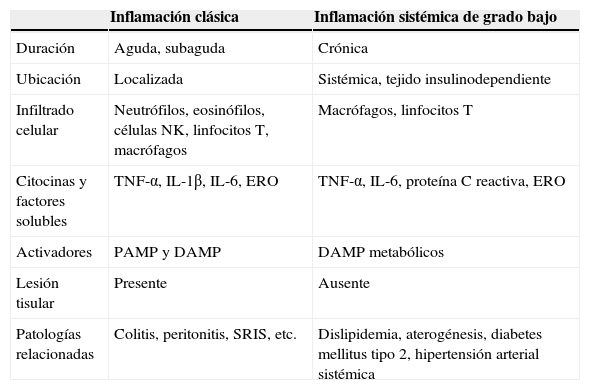
Evidencia reciente tanto experimental como clínica sugiere que el síndrome metabólico y la enfermedad cardiovascular podrían ser la consecuencia de un proceso inflamatorio sistémico1. Este cuadro de inflamación sistémica posee diferencias importantes con respecto a una respuesta inflamatoria clásica (tabla 1). De manera específica, la inflamación sistémica es caracterizada por una elevación en los niveles circulantes de proteínas de fase aguda y citocinas con actividad inflamatoria, tales como la proteína C reactiva (pCr), el factor de necrosis tumoral alfa (TNF-α) y las interleucinas (IL) 1β, 6 y 17, así como aumento en la infiltración de células inmunes como macrófagos y linfocitos T en tejido insulinodependiente2,3. Además, múltiples líneas de evidencia concuerdan en que la inflamación sistémica no induce lesión en el tejido inmunológicamente infiltrado, rasgo distintivo por el que se le ha acuñado el término de inflamación sistémica de grado bajo3,4. En otras palabras, durante un cuadro de inflamación sistémica de grado bajo el tejido exhibe niveles altos de factores inflamatorios y células inmunes infiltradas y, al mismo tiempo, no muestra alteraciones estructurales o pérdida en sus funciones primarias. Por otro lado, debido a su estrecha relación con el desarrollo de disfunciones cardiometabólicas en el paciente con obesidad, la inflamación sistémica de grado bajo ha recibido recientemente el nombre de metainflamación o inflamación metabólica5.
Principales diferencias entre la inflamación clásica y la inflamación sistémica de grado bajo
| Inflamación clásica | Inflamación sistémica de grado bajo | |
|---|---|---|
| Duración | Aguda, subaguda | Crónica |
| Ubicación | Localizada | Sistémica, tejido insulinodependiente |
| Infiltrado celular | Neutrófilos, eosinófilos, células NK, linfocitos T, macrófagos | Macrófagos, linfocitos T |
| Citocinas y factores solubles | TNF-α, IL-1β, IL-6, ERO | TNF-α, IL-6, proteína C reactiva, ERO |
| Activadores | PAMP y DAMP | DAMP metabólicos |
| Lesión tisular | Presente | Ausente |
| Patologías relacionadas | Colitis, peritonitis, SRIS, etc. | Dislipidemia, aterogénesis, diabetes mellitus tipo 2, hipertensión arterial sistémica |
DAMP: patrones moleculares asociados a daño; ERO: especies reactivas del oxígeno; NK: células asesinas naturales; IL: interleucina; PAMP: patrones moleculares asociados a patógenos; SRIS: síndrome de respuesta inflamatoria sistémica;TNF: factor de necrosis tumoral.
Sin embargo, a pesar de que el estudio de la relación entre la inflamación sistémica de grado bajo y el desarrollo de enfermedades cardiometabólicas ha aumentado considerablemente en la última década, aún existen numerosos elementos que son materia de investigación y debate en torno a esta. Por ejemplo, ¿cuáles son los mecanismos celulares que promueven el inicio y la perpetuación de la metainflamación?, ¿cuáles son los eventos moleculares involucrados en la manifestación clínica de la inflamación sistémica?, ¿existe evidencia que sugiera que padecimientos tales como la resistencia a la insulina, la dislipidemia, la hipertensión arterial y la diabetes mellitus tipo 2 tienen un trasfondo en la inflamación sistémica de grado bajo?
En esta revisión abordamos cada uno de los puntos anteriores, exponiendo la información más reciente con respecto a la inflamación sistémica de grado bajo y su relación con el desarrollo de alteraciones cardiometabólicas desde el punto de vista molecular y clínico, con un énfasis especial en la aplicación de este conocimiento en la identificación de posibles biomarcadores y blancos terapéuticos que redunden en mejores esquemas de tratamiento del paciente con alteraciones metabólicas.
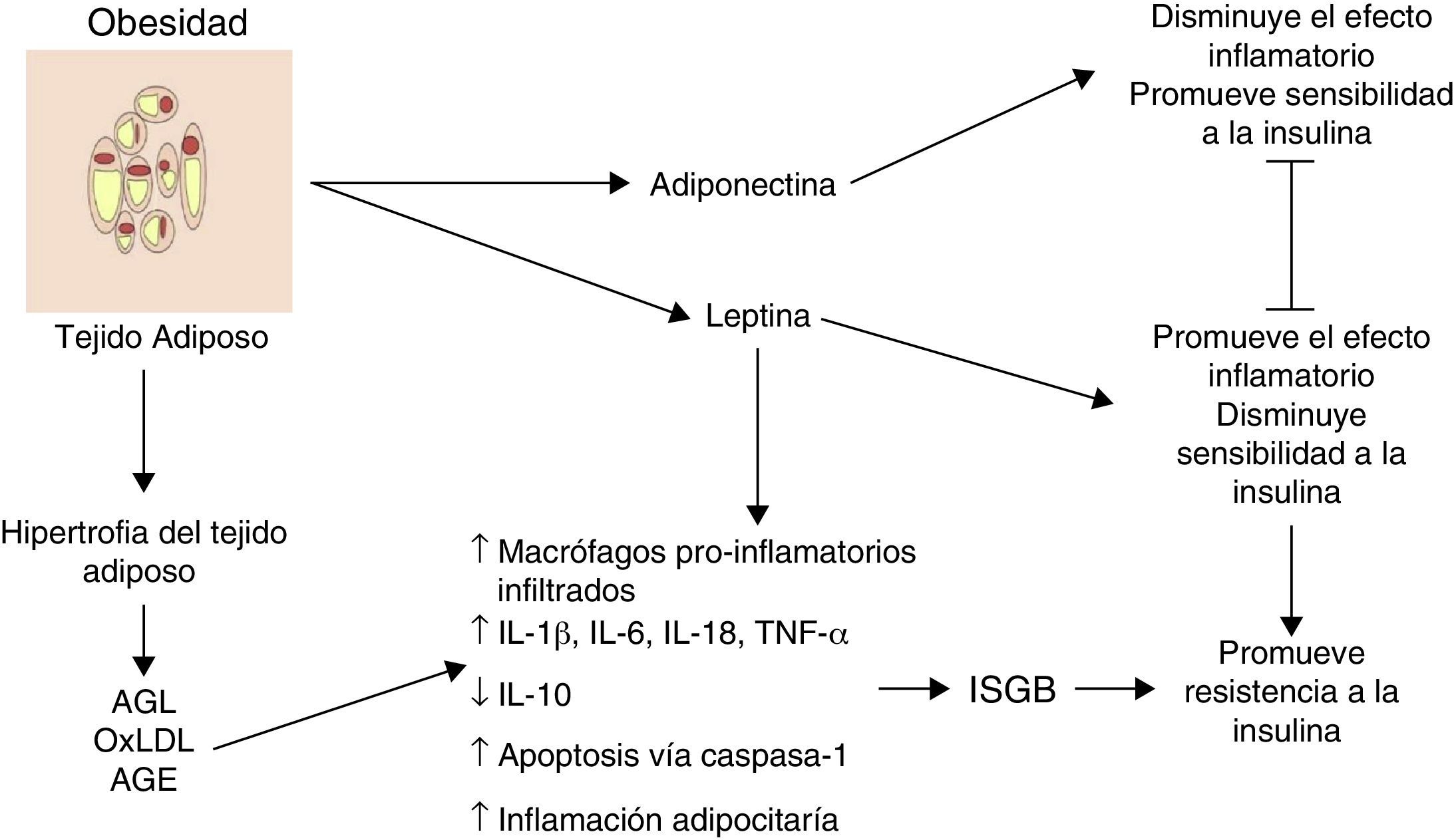
Inflamación del tejido adiposo visceral: el inicio de la inflamación sistémica de grado bajoUno de los primeros mecanismos implicados en el inicio de la inflamación sistémica de grado bajo es la inflamación en el tejido adiposo blanco o visceral (fig. 1). Como consecuencia del desbalance entre el consumo y el gasto energético, los adipocitos tienden a acumular grandes cantidades de ácidos grasos en su interior, lo cual conduce a procesos expansivos del tejido adiposo blanco como la hiperplasia y la hipertrofia adipocitaria (aumento en número y tamaño, respectivamente). En respuesta a modificaciones en el espaciamiento de la fracción vascular-estromal del tejido adiposo visceral, algunos adipocitos localizados en zonas lejanas a los vasos sanguíneos sufren hipoxia y posteriormente necrosis, tras lo cual son rodeados por células fagocíticas que inician un proceso inflamatorio orientado a la remoción de esas células6. Por otro lado, la gran cantidad de ácidos grasos almacenados en estas células es capaz de exacerbar procesos oxidativos como la lipoperoxidación, consistente en la oxidación de moléculas lipídicas al interior del adipocito. La lipoperoxidación que ocurre durante la hiperplasia/hipertrofia adipocitaria que conlleva a un escenario de estrés oxidativo celular caracterizado por un aumento considerable en los niveles de especies reactivas del oxígeno y el nitrógeno, tales como el ión superóxido (O2−) y el óxido nítrico (ON) respectivamente. Como consecuencia de este estallido oxidativo, numerosas células inmunológicas son reclutadas desde la periferia hacia el tejido adiposo, iniciando un proceso inflamatorio a nivel local caracterizado por elevación en los niveles de TNF-α y leptina, así como disminución en IL-10 y adiponectina6. Por otro lado, bajo condiciones de estrés como la hipoxia y la hiperoxidación de ácidos grasos, los adipocitos muestran alteraciones funcionales caracterizadas por un marcado estrés reticular asociado a procesos de plegamiento incorrecto de proteínas y autofagia, lo cual es capaz de desencadenar apoptosis en estas células7. Aunado a los eventos celulares descritos, este proceso apoptótico converge en un punto medular en el inicio de la inflamación sistémica de grado bajo, la inflamación del tejido adiposo blanco.

La inflamación del tejido adiposo visceral es una señal detonante en el inicio y la propagación de la inflamación sistémica de grado bajo.
AGE: productos terminales de glucosilación avanzada; AGL: ácidos grasos libres; IL: interleucina; ISGB: inflamación sistémica de grado bajo; oxLDL: lipoproteínas de baja densidad oxidadas; TNF: factor de necrosis tumoral;
Histológicamente, la inflamación del tejido adiposo es caracterizada por un infiltrado compuesto principalmente por macrófagos y secundariamente linfocitos T citotóxicos, con ausencia o muy poca presencia de neutrófilos3,8. Los macrófagos rodean a los adipocitos formando estructuras parecidas a corona (CLS, por sus siglas en inglés, de crown-like structures); un rasgo distintivo de la inflamación de grado bajo en el tejido adiposo6. Es importante resaltar que la infiltración de células inmunes es per se una fuente permanente de citocinas y factores proinflamatorios en el tejido adiposo hiperplásico e hipertrófico, el cual exhibe niveles elevados de IL-1β, IL-6, TNF-α y leptina, así como una mayor producción de proteínas con actividad quimio-atrayente o quimiocinas9. De hecho, el tejido adiposo hiperplásico e hipertrófico expresa concentraciones altas de quimiocinas como la proteína quimio-atrayente de macrófagos (MCP-1), el factor inhibitorio de la migración de macrófagos (MIF-1) y RANTES, todas estas con capacidad de atraer más macrófagos y linfocitos periféricos, perpetuando así el proceso de invasión inmune al tejido. Por otra parte, un cuerpo creciente de evidencia sugiere que los cambios a nivel del tejido adiposo visceral hiperplásico e hipertrófico no solo podrían tener una afectación autocrina y paracrina sino también endocrina, debido a que en sujetos con distintos grados de obesidad se observan niveles significativamente aumentados de citocinas, quimiocinas y monocitos-macrófagos inflamatorios en circulación1,2,10.
Por lo tanto, durante la obesidad la inflamación del tejido adiposo visceral podría desencadenar un proceso de inflamación sistémica, el mismo que afecta otros tejidos insulinodependientes como el hepático y muscular que perpetúa el estado inflamatorio a nivel adipocitario. Hasta ahora, hemos descrito el panorama celular que subyace a la inflamación del tejido adiposo visceral y su posible contribución al desencadenamiento de la inflamación sistémica de grado bajo. Sin embargo, ¿cuáles son los mecanismos moleculares implicados en la polarización del macrófago hacia el perfil inflamatorio con la consecuente liberación de TNF-α y el establecimiento de la inflamación sistémica de grado bajo? Esta cuestión será abordada a continuación.
El inicio de la inflamación sistémica de grado bajo depende de la activación de receptores de reconocimiento de patronesLas células inmunológicas, como los macrófagos y las células dendríticas, expresan un conjunto de receptores de membrana que forman parte de la respuesta innata ante estímulos externos e internos conocidos como receptores de reconocimiento de patrones (PRR, de Pattern Recognition Receptor). En general, los PRR son capaces de detectar dos tipos de moléculas, los patrones moleculares asociados a patógenos (PAMP, de pathogen-associated molecular patterns) y los patrones moleculares asociados a daño (DAMP, de damage-associated molecular patterns). Los PAMP son moléculas derivadas de microorganismos patógenos, mientras que los DAMP son liberados por tejidos que han sufrido algún tipo de daño. La interacción de los PRR con los PAMP o DAMP permite iniciar con rapidez una respuesta frente a procesos infecciosos o de daño tisular respectivamente, a través de la activación de la respuesta inflamatoria y/o de reparación del tejido. Es importante mencionar que el término PAMP está sujeto a debate actualmente, ya que microorganismos no patógenos también poseen muchas de estas estructuras moleculares y son capaces de inducir una respuesta inmune, por ejemplo: la respuesta de tolerancia inmunológica frente a la microbiota intestinal normal11.
Dentro de los diferentes grupos de PRR, el grupo integrado por los receptores tipo toll (TLR, por sus siglas en inglés, de toll-like receptor) ha sido caracterizado ampliamente como un conjunto de receptores cruciales para el reconocimiento tanto de PAMP como de DAMP12. En los seres humanos se han descrito 10 tipos funcionales de TLR, los cuales pueden estar localizados ya sea a nivel de la membrana celular o del citoplasma. Al unirse a sus ligandos, los TLR desencadenan una cascada de señalización intracelular que culmina con la activación de la respuesta inflamatoria a través de la secreción de mediadores inflamatorios y quimiotácticos que reclutan células inmunológicas a los tejidos infectados o dañados. Si bien el papel de los TLR ha sido ampliamente descrito en procesos infecciosos, un cuerpo creciente de evidencia sugiere que estos receptores podrían desempeñar un papel clave durante el inicio y la perpetuación de la inflamación sistémica de grado bajo asociada con la obesidad13.
A diferencia de la inflamación aguda o clásica inducida por PAMP, la respuesta asociada con diferentes moléculas derivadas de un estado metabólico alterado o «DAMP metabólicos» conduce a una inflamación de menor intensidad, mayor duración y sistémica14. De esta forma, distintos metabolitos han sido vinculados como generadores de inflamación sistémica de grado bajo a través de su unión a TLR14. En este sentido, tanto la dislipidemia como la hiperglucemia han mostrado participar decisivamente en la polarización de macrófagos hacia un fenotipo proinflamatorio y el inicio concomitante de la inflamación sistémica de grado bajo por medio de la activación de los TLR. De manera específica, se sabe desde hace tiempo que los ácidos grasos libres (AGL), sobre todo los saturados como el ácido palmítico, generan una respuesta inflamatoria sistémica12. Actualmente sabemos que este fenómeno se presenta debido a que durante la dislipidemia la gran cantidad de AGL son capaces de unirse a TLR-1, 2 y 6 localizados en la superficie del macrófago, formando heterodímeros que unen lipopéptidos di- o triacilados e induciendo la liberación de citocinas inflamatorias15.
Por otro lado, además de reconocer al lipopolisacárido (LPS, una molécula derivada de bacterias gramnegativas), el TLR-4 es también capaz de reconocer proteínas séricas glucosiladas denominadas productos terminales de glucosilación avanzada (AGE, de advanced glycation end-products)16. Durante un estado hiperglucémico diversas proteínas como la hemoglobina, la albúmina o las lipoproteínas de baja densidad (LDL) son glucosiladas de manera no enzimática en residuos de lisina y arginina dando origen a los AGE. En células como los macrófagos, estos AGE pueden unirse al TLR-4, el cual se encuentra sobreexpresado en enfermedades metabólicas13. La activación de TLR conduce a una cascada de señalización intracelular mediada por el factor nuclear kappa B (NF-κB), el cual una vez translocado al núcleo activa la transcripción de genes que codifican para citocinas y quimiocinas inflamatorias, tales como IL-1β, TNF-α y CXCL8, promoviendo así el inicio de una respuesta inflamatoria en respuesta a la hiperglucemia.
Por lo tanto, estados metabólicos alterados como la dislipidemia y la hiperglucemia pueden inducir una respuesta inflamatoria en el macrófago a través de la activación por PRR, la misma que podría extenderse posteriormente a nivel sistémico cuando esta célula migre hacia tejido insulinodependiente y altere el microambiente de citocinas presente en el organismo. Por lo tanto, derivado de estos estudios una de las posibles estrategias terapéuticas encaminadas en el control de la inflamación sistémica de grado bajo en el paciente obeso implica el diseño de fármacos que impidan la unión de AGL y AGE con los TLR-1, 2, 4 y 6, o bloqueen la activación de mediadores celulares como NF-κB, inhibiendo potencialmente el inicio y progreso de la inflamación sistémica y sus consecuencias metabólicas. Sin embargo, la relevancia clínica de este tipo de estrategias novedosas aún está por ser determinada.
La información revisada hasta el momento nos permite conocer con mayor detalle los mecanismos celulares y moleculares implicados en el establecimiento de la inflamación sistémica de grado bajo durante la obesidad. Sin embargo, surgen a este nivel un par de preguntas que serán materia de interés en el resto de este documento, ¿cuáles son la manifestaciones clínicas del inicio y la perpetuación de la inflamación sistémica de grado bajo en el sujeto obeso?, ¿existe evidencia clínica de la relación entre la inflamación sistémica de grado bajo y el desarrollo de aterogénesis, diabetes mellitus tipo 2 e hipertensión arterial sistémica? La respuesta a estas cuestiones será abordada a continuación.
Dislipidemia, aterogénesis e inflamación sistémica de grado bajoUn número creciente de evidencia ha sugerido recientemente que la aterogénesis podría ser la consecuencia de un proceso metainflamatorio de afectación vascular, en el cual la formación de una placa obstruye parcial o totalmente la luz arterial llevando a eventos como el infarto de miocardio, la enfermedad vascular cerebral o la muerte súbita17. Junto con la inflamación sistémica de grado bajo, la dislipidemia ha sido identificada como un factor decisivo en la aterogénesis y en los riesgos vasculares que esta implica17. En la placa naciente, las LDL oxidadas (oxLDL) se unen a receptores que se encuentran en la superficie de macrófagos, tales como CD36, TLR-4, el receptor de productos terminales de glucosilación avanzada (RAGE, de receptor for advanced glycation end-products) y el receptor del péptido formilado 2 (FPR2, de formyl-peptide receptor 2)18–20. Esta interacción induce en el macrófago la liberación de ERO, las mismas que activan el inflamasoma NLRP3 (de nucleotide-binding oligomerization domain-like receptor family of proteins), un complejo de proteínas con actividad inflamatoria y apoptótica en respuesta a PAMP y DAMP21. En el mismo sentido, se ha observado que los AGL son capaces de inducir directamente liberación de agentes inflamatorios (TNF-α e IL-1β) y oxidantes (ON) en la célula endotelial al inducir desacoplamiento mitocondrial independiente del inflamasoma22. Esta cascada de eventos inflamatorios lleva al endotelio vascular a la liberación de moléculas de adhesión como la molécula de adhesión intracelular (ICAM-1, de intracelular adhesión molecule 1) y la molécula de adhesión celular vascular (VCAM-1, de vascular cell adhesión molecule 1), las mismas que favorecen el direccionamiento de nuevos macrófagos de la periferia hacia el endotelio y subendotelio vascular. Debido a la captación de LDL, los macrófagos invasores del espacio subendotelial se transforman en células espumosas, evento que junto con la migración de células musculares con alta capacidad de expresar TNF-α, IFN-γ, IL-1β, e IL-6, es fundamental en el engrosamiento de la placa ateroesclerótica y el aumento del riesgo cardiovascular del paciente23. Por otro lado, no solo las oxLDL contribuyen a la aterosclerosis, numerosos lípidos y fosfolípidos oxidados han sido identificados en las placas ateroscleróticas, entre los cuales destacan el factor activador de plaquetas (PAF, por sus siglas en ingles, de platelet activating factor), los fosfolípidos oxidados y la lisofosfatidilcolina, que por sí mismos son capaces de inducir la expresión de TNF-α, IL-1β, IL-6, TLR-2, TLR-4 y NF-κB en macrófagos, y de MCP-1 y VCAM en células endoteliales23–25. Además, otras moléculas como la pCr pueden depositarse en la íntima arterial y reclutar a través de receptores de baja afinidad para IgG monocitos-macrófagos periféricos con actividad inflamatoria26. En el mismo sentido, diversos estudios sugieren que la IL-6 podría desempeñar un papel importante como reguladora de la lipidemia, ya que el bloqueo en la función de este mediador inflamatorio por medio de tocilizumab mejora la relación entre el colesterol total y las lipoproteínas de alta densidad (HDL)27. Con base en toda esta evidencia, actualmente se considera a la alteración lipídica como una inductora fundamental de metainflamación en la aterogénesis, ya que: 1) las LDL y oxLDL son estímulos netamente inflamatorios, 2) la aterosclerosis no se desarrolla en modelos animales o en pacientes en donde no existe un nivel significativo de estas moléculas, y 3) múltiples moléculas netamente inflamatorias como pCr e IL-6 pueden regular el proceso ateroesclerótico27. En esta aparente relación dislipidemia –inflamación sistémica de grado bajo– aterogénesis, uno de los tópicos de mayor interés por su posible aplicación médica es el valor pronóstico de los anticuerpos anti-oxLDL en la aterosclerosis, ya que pacientes con enfermedad coronaria muestran disminución en los anticuerpos anti-oxLDL asociada con mayor severidad del síndrome isquémico agudo28. De forma complementaria, la infusión de anticuerpos anti-oxLDL disminuye la aterogénesis en ratones susceptibles al desarrollo de ateroesclerosis29. Sin embargo, es necesario incluir la determinación del título de estos anticuerpos en cohortes prospectivas de pacientes para conocer su verdadero valor pronóstico y comprender con mayor profundidad el papel de la respuesta inmune en la aterogénesis.
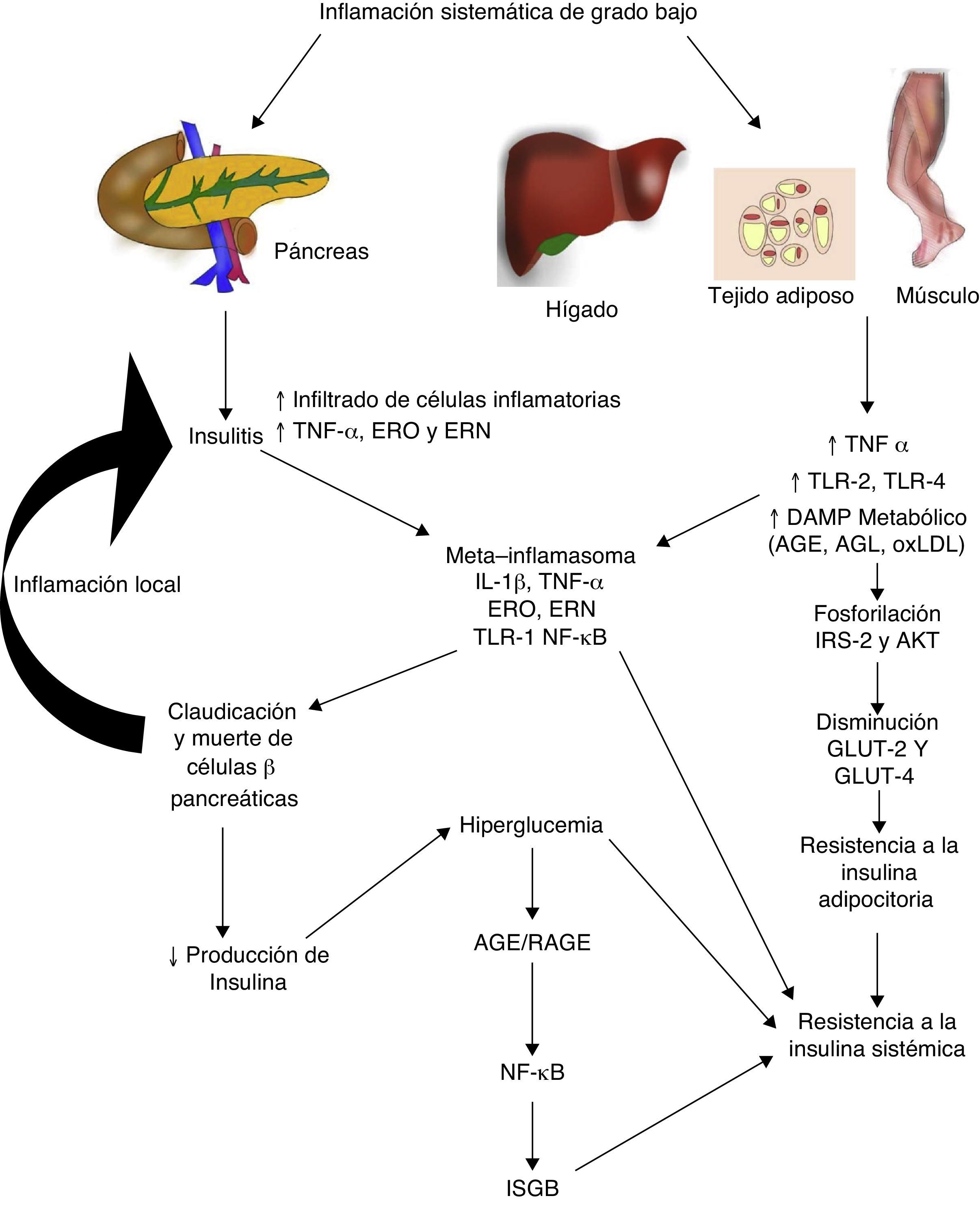
Diabetes mellitus tipo 2 e inflamación sistémica de grado bajoLa diabetes mellitus tipo 2 es una enfermedad de etiología multifactorial caracterizada por hiperglucemia crónica asociada con resistencia a la acción de la insulina y una respuesta compensatoria inadecuada en la secreción de esta hormona. Numerosos estudios indican que la fisiopatogenia de esta enfermedad está íntimamente relacionada con el proceso inflamatorio sistémico, el mismo que podría estar activo antes de que se desarrollen las alteraciones metabólicas detectables clínicamente (fig. 2)30. Inclusive, los hijos de personas con diabetes mellitus tipo 2 presentan elevación de marcadores inflamatorios mucho antes de que presenten alguna alteración metabólica31.

La inflamación sistémica de grado bajo contribuye al desarrollo de la resistencia a la insulina por distintas vías.
AGE: productos terminales de glucosilación avanzada; AGL: ácidos grasos libres; AKT: proteína cinasa B; ERN: especies reactivas del nitrógeno; ERO: especies reactivas del oxígeno; GLUT: proteínas transportadoras de glucosa; IL: interleucina; ISGB: inflamación sistémica de grado bajo; IRS: sustrato del receptor de insulina; NF-κβ: factor nuclear kappa beta; oxLDL: lipoproteínas de baja densidad oxidadas; RAGE: receptor de productos terminales de glucosilación avanzada; TLR: receptor tipo toll; TNF: factor de necrosis tumoral.
En la diabetes mellitus tipo 2 la inflamación del islote pancreático juega un papel primordial. El proceso inflamatorio en la célula β-pancreática es mediado por dos vías: 1) la activación de TLR-2 y TLR-4 por DAMP metabólicos como los AGE, y 2) el ensamblaje del inflamosoma NLRP332. El resultado de este proceso es la insulitis caracterizada por una liberación continua de IL-6, IL-8, TNF-α y MCP-1, activación de macrófagos insulares y reclutamiento de nuevos monocitos-macrófagos periféricos. Otras moléculas como la IL-12, IL-17 y la NADPH oxidasa-1 tienen también un papel importante en la inflamación de los islotes32. En la insulitis, la célula β-pancreática es la más susceptible a los efectos de IL-1β, ya que expresa mayores niveles del receptor de esta citocina inflamatoria (IL-1R1)33. Como nuestro grupo predijo en 201134, el bloqueo de los efectos de IL-1β con el anticuerpo monoclonal Gevokizumab resultó en mejoría en la secreción del péptido C, disminuyendo los niveles de hemoglobina glucosilada y citocinas inflamatorias en pacientes con diabetes mellitus tipo 235.
Por otro lado, este proceso inflamatorio afecta simultáneamente a otros tejidos a través de una cascada de eventos que constituye actualmente una de las principales teorías fisiopatogénicas en el desarrollo de resistencia a la insulina y diabetes mellitus tipo 2. Durante la inflamación sistémica de grado bajo, el tejido adiposo, hepático y muscular sufre infiltración de macrófagos inflamatorios productores de TNF-α, citocina que interfiere directamente con la capacidad de estos tejidos de responder a la insulina. En condiciones normales, la insulina se une a su receptor localizado en la superficie de adipocitos, hepatocitos y miocitos, iniciando una cascada de señalización intracelular mediada por el sustrato del receptor de insulina (IRS, de insulin receptor substrate), el mismo que activa AKT y finalmente induce la movilización de proteínas transportadoras de glucosa (GLUT-2 y 4) para la incorporación de este azúcar al interior de la célula. En presencia de TNF-α, la activación de la vía de señalización IRS-AKT-GLUT es inhibida por la acción de vías intracelulares responsivas a TNF-α como ERK/JNK, PTP1B y NF-κB, las mismas que compiten o inhiben directamente la fosforilación de IRS-2, AKT, GLUT-2 y 436. En síntesis, este escenario inflamatorio conduce a un estado de hiperglucemia y posteriormente uno compensatorio de hiperinsulinemia, seguido por la claudicación y apoptosis de la célula β-pancréatica, insulitis, disminución en los niveles de insulina y el establecimiento de la diabetes mellitus tipo 2. Como mencionamos anteriormente, la hiperglucemia conlleva per se a un estado de inflamación sistémica caracterizado por activación de macrófagos infiltradores de tejido y, de manera concomitante, elevación de citocinas inflamatorias, eventos que permiten la perpetuación de la inflamación sistémica de grado bajo y sus consecuencias a nivel metabólico.
La hipertensión arterial como resultado de múltiples mecanismos inflamatoriosComo entidad clínica, la hipertensión arterial sistémica se manifiesta entre los 20 y 50 años de edad, con prevalencias hasta del 75% en edades avanzadas37. La hipertensión arterial sistémica está estrechamente relacionada con padecimientos tales como: la enfermedad vascular cerebral, la enfermedad coronaria, la insuficiencia cardiaca, la fibrilación auricular y, la insuficiencia renal. Existen diferentes mecanismos moleculares que convergen en el desarrollo del fenotipo clínico de hipertensión arterial sistémica en los cuales la inflamación parece desempeñar un papel central en el engrosamiento de las capas de la íntima y media, así como en la rigidez de las arterias y de manera muy importante en la disfunción endotelial.
Cambios en el perfil de expresión del endotelio vascular se han relacionado con mayor adherencia y aterogenicidad. Estos cambios incluyen la secreción de moléculas como selectina endotelial soluble, trombomodulina, VCAM-1, ICAM-1 y factor de von Willebrand31,32, y se dan como consecuencia del estímulo de diversos DAMP metabólicos como los FFA, especialmente los ácidos grasos insaturados. Estas moléculas tienen un efecto negativo en la función endotelial al activar NF-κB e IL-6 y promover la producción de ERO32. Una de las moléculas inflamatorias mayormente implicadas en el desarrollo de hipertensión arterial sistémica ha sido la pCr38. En un estudio que incluyó a 320 pacientes no diabéticos, con reciente diagnóstico de hipertensión en quienes se determinó el nivel de pCr, RAGE soluble (sRAGE) y dimetilarginina asimétrica, se observó que los niveles más altos de sRAGE correlacionaban con mayor índice de masa corporal, mayor grado de hipertensión y mayores niveles de pCr39. La pCr disminuye la producción de ON por su acción sobre la sintasa de ON endotelial (eNOS) e induce mayor expresión de receptores tipo 1 de angiotensina (ATR1) en la musculatura vascular lisa, favoreciendo la activación de NF-κB por angiotensina II y la liberación de TNF-α, señales que inciden negativamente sobre la eNOS40. Es importante señalar que la pCr inhibe también a la sintasa de prostaciclina, por lo que interfiere con el efecto vasodilatador mediado por prostaciclinas41. En células endoteliales humanas aórticas, la pCr inhibe la GTP ciclohidrolasa, disminuyendo la síntesis de tetrahidrobiopterina (BH4, cofactor de la eNOS) y estimulando a la NADPH oxidasa, lo que incrementa los niveles de ERO. El efecto final es la reducción en la producción de ON y con ello, hipertensión. En sujetos con descontrol hipertensivo matutino, el tener pCr sérica elevada incrementa el riesgo cardiovascular 5.77 veces (IC 95% 2.11-15.81) en comparación con los que tienen un nivel normal de pCr42. En comparación con los pacientes con hipertensión arterial sistémica sin descontrol matutino, las placas de ateroma de aquellos con descontrol matutino tienen mayor actividad inflamatoria, caracterizada por mayor cantidad de macrófagos y linfocitos T infiltrados, así como producción de NF-κB y TNF-α. Este fenotipo inflamatorio se asocia con mayor actividad del ubiquitina-proteasoma, que es la vía intracelular principal no lisosomal de degradación de proteínas que han sido marcadas para su degradación por proteasoma43. Esta vía es necesaria para la activación del NF-κB ya que degrada las proteínas inhibitorias IKK (por sus siglas en inglés, de inhibitor of NF-κB kinase). En modelos animales de hipertensión se determinó que la infiltración de tejidos por macrófagos y la producción de NF-κB e IL-1β es más intensa en el riñón, lo que favorece la activación del sistema renina-angiotensina-aldosterona y participa en el daño renal inducido por los hipertensión44.
Por lo tanto, el reconocimiento de la hipertensión arterial sistémica como un proceso inflamatorio sistémico es fundamental no solo para entender la génesis de la enfermedad sino también el desarrollo de complicaciones asociadas con esta. Por esta razón, el abordaje de tratamiento desde un punto de vista inmunológico podría tener beneficios para el paciente. Por ejemplo: en pacientes con insuficiencia cardiaca secundaria a hipertensión arterial sistémica, los efectos benéficos del carvedilol son menores en quienes tienen niveles mayores de citocinas inflamatorias como TNF-α e IL-1845. En síntesis, la inflamación sistémica de grado bajo no solo participa en el desarrollo de hipertensión arterial sistémica sino que también se asocia con su gravedad y toma parte en el desarrollo de complicaciones como la hipertrofia y disfunción ventricular izquierda, la arritmogénesis y la aterosclerosis, por lo que el estudio de esta es fundamental para llegar a un mejor entendimiento de la enfermedad y lograr un abordaje óptimo en el paciente.
ConclusionesLa evidencia presentada nos indica que la inflamación sistémica de grado bajo juega un papel fundamental en el desarrollo de trastornos metabólicos, tales como las dislipidemias, la aterogénesis, la diabetes mellitus tipo 2 y la hipertensión arterial sistémica. Esta metainflamación conduce a la activación de señales intracelulares que culminan con la liberación de factores inflamatorios de afectación local y sistémica, los cuales retroalimentan e interconectan a los tejidos adiposo, muscular y hepático con macrófagos proinflamatorios productores de citocinas inflamatorias y ERO. Una vez establecida, la inflamación sistémica de grado bajo promueve y perpetúa las alteraciones metabólicas estableciendo un ciclo deletéreo que favorece procesos patológicos como la resistencia a la insulina, la arterioesclerosis y la disfunción endotelial. La rotura de este ciclo depende entonces de controlar simultáneamente tanto el componente metabólico como el inflamatorio. Es por eso que en el futuro próximo, la investigación se deberá orientar hacia el diseño de terapias antiinflamatorias que favorezcan una adecuada regulación metabólica, tomando en cuenta el nivel de citocinas y células inmunes circulantes, factores de transcripción y adhesión, así como el perfil de DAMP metabólicos en el paciente. Junto con la clínica, este abordaje más integral podría contribuir a identificar diferentes fenotipos de metainflamación dependiendo del grado y la relación entre sus componentes, los cuales podrían ser identificados por el clínico a través de biomarcadores que permitan predecir la evolución de un paciente con enfermedad metabólica y establecer terapias antiinflamatorias personalizadas basadas en la evidencia molecular.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos la lectura crítica del presente manuscrito y la asistencia técnica en el diseño de los cuadros a la bióloga experimental Angélica Fabiola Barragán.