



La heredabilidad y la penetrancia familiar de los trastornos causantes de la parada cardíaca en ausencia de cardiopatía aparente es desconocida. Nuestro objetivo es describir los hallazgos clínicos y los medios para alcanzar el diagnóstico en una muestra de familiares de probandos con parada cardíaca en ausencia de cardiopatía aparente.
Material y métodosEstudiamos a familiares de primer a tercer grado de pacientes afectados por parada cardíaca de origen incierto. En todos se realizó electrocardiograma y ecocardiograma transtorácico. En los casos con diagnóstico cierto en el probando, se realizó además estudio dirigido en función del mismo para desenmascarar la enfermedad.
ResultadosSe incluyó a 88 sujetos pertenecientes a 35 familias (media de 2,5 familiares por caso índice, con una edad media de 38,4 años y 52,8% varones). El 55,6% fueron familiares de primer grado y el resto de segundo y tercer grado. En 18 de los 35 casos índice se alcanzó un diagnóstico final. Se obtuvo un diagnóstico positivo en 19 familiares (21,5%) consistentes en síndrome de Brugada (10 casos), síndrome de QT largo (3 casos), taquicardia ventricular catecolaminérgica polimórfica (5 casos) y un caso de posible Síndrome de QT corto. En 4 familias, el diagnóstico del probando se alcanzó exclusivamente mediante el estudio familiar.
ConclusionesLa prevalencia de canalopatías cardíacas entre los familiares de probandos con parada cardíaca de origen incierto fue alta, con frecuente necesidad de test de desenmascaramiento y genético para logar un diagnóstico cierto.
Familial inheritance and penetrance of disorders causing unexplained cardiac arrest is not well known. Our aim is to describe clinical and diagnostic methods in a sample of unexplained cardiac arrest relatives.
Material and methodsWe studied first to third degree relatives of unexplained cardiac arrest patients with electrocardiogram and echocardiogram. In all cases with a definitive diagnosis in the index case, phenotype directed unmasking tests were performed in all available relatives.
ResultsWe included 88 subjects belonging to 35 families (mean of 2.5 relatives per family), with a mean age of 38,4 years, and 52,8% males. 55,6% were first degree relatives, and the remaining were second and third degree. A definitive diagnosis was achieved in 19 relatives (21,5%), consisting of Brugada Syndrome (10 cases), long QT syndrome (3 cases), catecholaminergic polymorphic ventricular tachycardia (5 cases) and one case of possible short QT syndrome. In 4 families, the diagnosis in the proband was reached thanks to the familial assesment.
ConclusionsPrevalence of cardiac channelopathies among relatives of unexplained cardiac arrest patients was high, using a multitest approach including unmasking pharmacological tests and genetic study.
La parada cardíaca (PC) de origen incierto es una entidad infrecuente debida principalmente a canalopatías cardíacas y miocardiopatías con baja penetrancia clínica1-3. Entre estas se encuentran fundamentalmente el síndrome de QT largo (SQTL) y corto, el síndrome de Brugada (SB), la taquicardia ventricular catecolaminérgica (TVCP) y la displasia arritmogénica de ventrículo derecho. Estudios previos demuestran un significativo porcentaje de casos portadores de mutación patogénica, sobre todo de enfermedades eléctricas primarias, sin expresión en el electrocardiograma (ECG) pero sin que ello suponga estar libre de riesgo de arritmias ventriculares4-6. La mayoría de estos trastornos se transmiten de forma autosómica dominante, por lo que es esperable una alta prevalencia oculta entre los familiares.
Escasos estudios previos han analizado la penetrancia y la heredabilidad en las familias con un miembro afectado por PC de causa poco clara2,7-9. En el registro CASPER, estudio más amplio hasta la fecha en pacientes con PC idiopática, el porcentaje de familiares afectados fue del 24%1, pero no aportaba información acerca del protocolo ni test diagnósticos empleados en los mismos. El estudio familiar se recomienda actualmente como parte de la cascada diagnóstica tras un hallazgo positivo en el probando, pero no de manera sistemática a pesar de no hallar fenotipo en el probando10.
Nuestro objetivo es describir los hallazgos clínicos y los medios para alcanzar el diagnóstico en una muestra de familiares de probandos con PC en ausencia de cardiopatía aparente, tras la aplicación de un protocolo sistemático descrito por nuestro grupo3.
Material y métodosPoblación de estudioSe incluyó a familiares de primer a tercer grado de 35 pacientes pertenecientes a 9 centros españoles, afectados por una PC, y que estuviesen libres de enfermedad conocida en el momento del estudio. Definimos la PC de origen incierto en probandos reanimados de un paro cardíaco en FV o taquicardia ventricular (TV) polimórfica sin pulso sin causa aparente en el estudio convencional, en presencia de ECG no diagnóstico de enfermedad, con ecocardiograma sin hallazgos patológicos y coronariografía sin lesiones angiográficas mayores al 50% de estenosis. En todos los casos índice se aplicó el protocolo descrito por nuestro grupo en una publicación reciente, consistente en realización secuencial de test farmacológicos, estudio familiar y genético con next generation sequencing3,11. Se seleccionó a los familiares con mayor grado de parentesco con el probando y se les ofreció participar en el registro, previa firma del consentimiento informado.
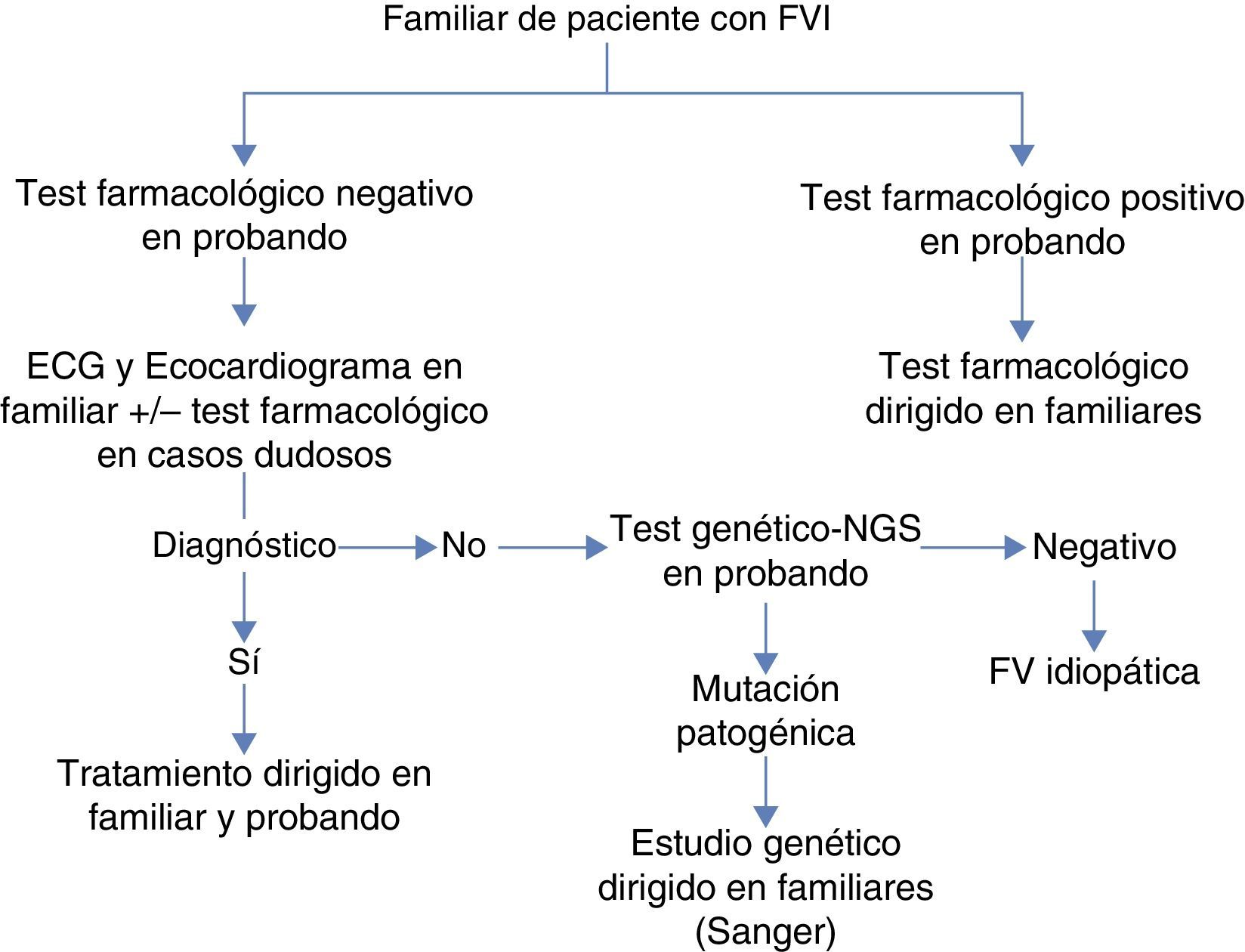
ProtocoloEl protocolo de estudio en familiares se aplicó tal y como se expone en la figura 1. En aquellos casos en que el diagnóstico de canalopatía se hubiese obtenido mediante test farmacológicos, las pruebas en los familiares fueron enfocadas al desenmascaramiento de la misma mediante ergometría o test de epinefrina para la TVCP y SQTL, y test de flecainida si el probando presentaba un SB. En los casos con negatividad en el probando en los test farmacológicos, se estudió se forma sistemática a los familiares con ECG y ecocardiograma transtorácico y en los casos dudosos se realizaba también test farmacológico. Finalmente, si el diagnóstico en el probando se llevó a cabo con la detección de una mutación genética causal, se procedió a secuenciar la misma mediante la técnica de Sanger en los familiares incluidos. Aquellos probandos en los que tras la aplicación del protocolo no se hubiese llegado al diagnóstico y en el ECG basal mostrasen un patrón de repolarización precoz fueron diagnosticados de síndrome de repolarización precoz10.

La ergometría se realizó según el protocolo estándar de Bruce dirigida por fenotipo o en caso de contraindicación para la realización de test de epinefrina. Primero se realizaba el test de epinefrina y, en caso de negatividad, tras un período de lavado se procedía al test de flecainida. Para el test de epinefrina se siguió el protocolo de Shimizu et al.12, con paso a través de vía intravenosa periférica de bolo inicial y posterior infusión continua durante 15 min de dilución de 1mg/0,1L a un ritmo de 0,1 μg/kg/min hasta 0,2 μg/kg/min. De acuerdo con Shimizu et al. y Khran et al.12,13, el test se consideró positivo para SQTL si se observaba prolongación del intervalo QT absoluto>30ms en presencia de taquicardización, o bien por aparición de muescas en la rama ascendente de la onda T, indicativas de SQTL tipo 2. Por el contrario, se diagnosticó TVCP si aparecían más de 3 extrasístoles polimórficos o TV bidireccional13. El estudio genético en familiares se realizó una vez demostrada una variante probablemente relacionada con la enfermedad en el probando. Se realizó mediante la técnica de Sanger®, y para definir una variante genética como patogénica y evitar de falsos positivos se exigió demostración previa en estudios clínicos o funcionales de causalidad, cosegregación del fenotipo en los miembros de la familia afectada, relación coherente del genotipo observado con el fenotipo presente en el probando (desencadenantes de arritmia, estudio familiar, pruebas de imagen, ECG, etc.) y predictores bioinformáticos de patogenicidad. En caso de no haber una evidencia sólida de causalidad, se interpretó como posible diagnóstico, pero no definitivo.
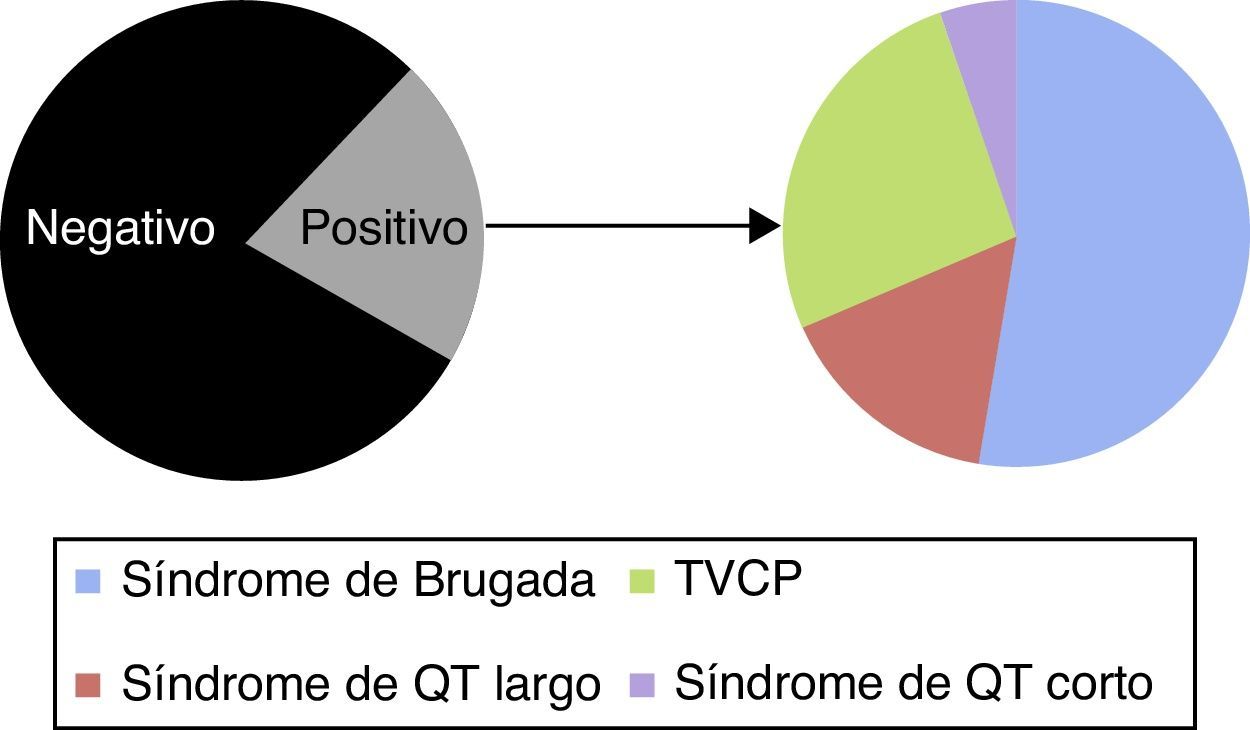
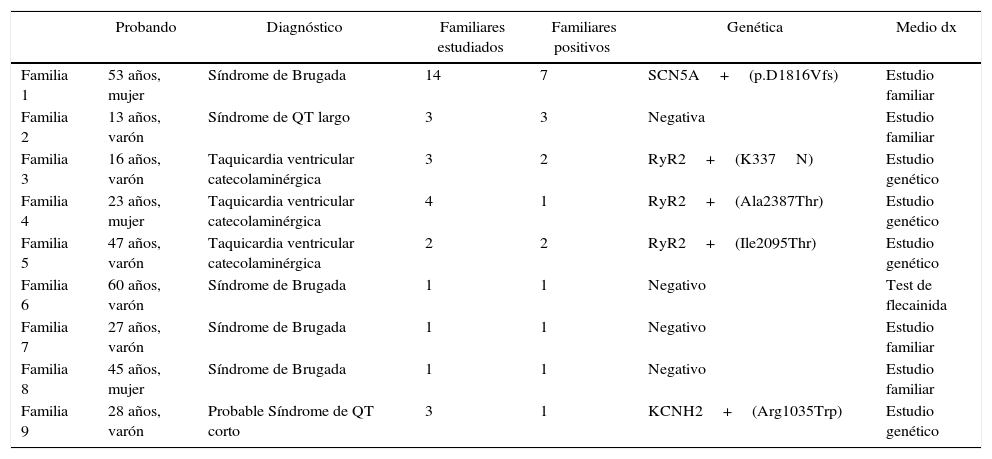
ResultadosSe incluyó a 88 familiares de primer (media de 2,5 familiares por caso índice), segundo o tercer grado pertenecientes a 35 familias donde hubo un caso de FVI. La edad media fue de 38,4 años y el 52,8% varones, siendo un 55,6% de primer grado, y los restantes de segundo o tercer grado. Se detectaron 19 casos de canalopatía ocultos entre los familiares (21,5%), siendo 11 de ellos en familiares de primer grado, quedando estas reflejadas en la figura 2. En 9 (25,7%) de las 35 familias estudiadas se encontró la canalopatía en algún miembro estudiado, y referido a los casos con probando con estudio positivo, este porcentaje ascendía al 50%. Los medios más frecuentes para obtener el diagnóstico fueron el test genético apoyado en la detección de una mutación causal en el probando, y el test farmacológico dirigido en familiares tras la detección de un ECG patológico basal. La tabla 1 muestra un resumen de las familias en las que se obtuvieron resultados positivos y en base a qué medio diagnóstico se realizó el diagnóstico, así como la información genética. El estudio familiar fue crucial para establecer el diagnóstico en 4 familias. Definimos diagnóstico en el probando por estudio familiar cuando el probando mostró en todo momento negatividad de los test convencionales y farmacológicos, y fue un hallazgo indicativo en el ECG de uno de los familiares directos el que motivó la realización del test de epinefrina o flecainida que finalmente arrojó el diagnóstico familiar. En resumen, en 4 casos el diagnóstico familiar se realizó mediante el estudio familiar, en 4 casos mediante test genético y en uno con test de provocación en el familiar directo.

Nueve familias con detección de miembros afectados
| Probando | Diagnóstico | Familiares estudiados | Familiares positivos | Genética | Medio dx | |
|---|---|---|---|---|---|---|
| Familia 1 | 53 años, mujer | Síndrome de Brugada | 14 | 7 | SCN5A+(p.D1816Vfs) | Estudio familiar |
| Familia 2 | 13 años, varón | Síndrome de QT largo | 3 | 3 | Negativa | Estudio familiar |
| Familia 3 | 16 años, varón | Taquicardia ventricular catecolaminérgica | 3 | 2 | RyR2+(K337N) | Estudio genético |
| Familia 4 | 23 años, mujer | Taquicardia ventricular catecolaminérgica | 4 | 1 | RyR2+(Ala2387Thr) | Estudio genético |
| Familia 5 | 47 años, varón | Taquicardia ventricular catecolaminérgica | 2 | 2 | RyR2+(Ile2095Thr) | Estudio genético |
| Familia 6 | 60 años, varón | Síndrome de Brugada | 1 | 1 | Negativo | Test de flecainida |
| Familia 7 | 27 años, varón | Síndrome de Brugada | 1 | 1 | Negativo | Estudio familiar |
| Familia 8 | 45 años, mujer | Síndrome de Brugada | 1 | 1 | Negativo | Estudio familiar |
| Familia 9 | 28 años, varón | Probable Síndrome de QT corto | 3 | 1 | KCNH2+(Arg1035Trp) | Estudio genético |
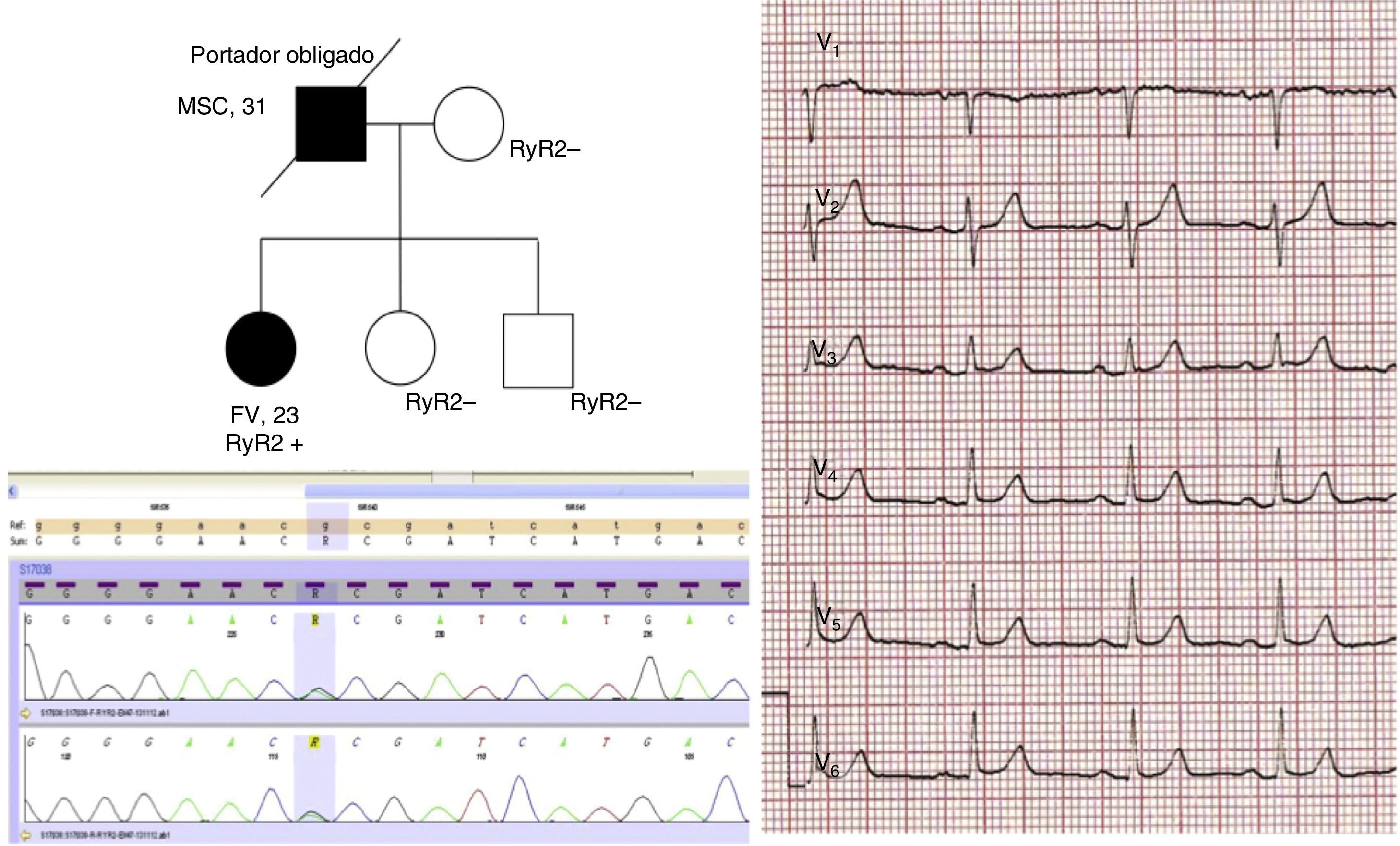
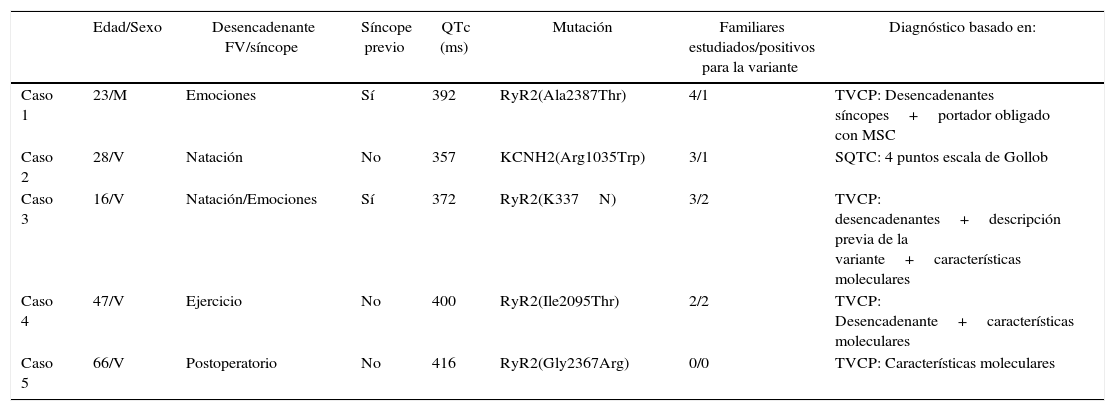
En conjunto, se alcanzó un diagnóstico final en 18 de los 35 probandos/familias (51,4%), siendo el 22,2% de estos a través del estudio familiar, ya que este fue crucial para establecer el diagnóstico en 4 familias. En ellas, el probando mostró en todo momento negatividad de los test convencionales y farmacológicos, y fue un hallazgo indicativo en el ECG de uno de los familiares directos el que motivó la realización del test de epinefrina o flecainida que finalmente arrojó el diagnóstico familiar. En otras 5 de las familias, el diagnóstico se realizó sobre la base exclusivamente del hallazgo de una mutación genética probablemente causal. Tal y como muestra la tabla 2, el estudio familiar en estos casos fue útil en algunas de las familias para establecer la cosegregación del hallazgo genético; sin embargo, en ocasiones tampoco el estudio familiar conseguía desenmascarar el fenotipo y hay casos con ausencia total de fenotipo que se diagnosticaron sobre la base de datos clínicos epidemiológicos apoyados en las características moleculares de la variante genética hallada. La figura 3 muestra un ejemplo ilustrativo de esto, con la detección de una mutación patogénica en la rianodina facilitando el diagnóstico de TVCP.
Diagnóstico en probando y familiares mediante estudio genético de 5 casos con fenotipo negativo
| Edad/Sexo | Desencadenante FV/síncope | Síncope previo | QTc (ms) | Mutación | Familiares estudiados/positivos para la variante | Diagnóstico basado en: | |
|---|---|---|---|---|---|---|---|
| Caso 1 | 23/M | Emociones | Sí | 392 | RyR2(Ala2387Thr) | 4/1 | TVCP: Desencadenantes síncopes+portador obligado con MSC |
| Caso 2 | 28/V | Natación | No | 357 | KCNH2(Arg1035Trp) | 3/1 | SQTC: 4 puntos escala de Gollob |
| Caso 3 | 16/V | Natación/Emociones | Sí | 372 | RyR2(K337N) | 3/2 | TVCP: desencadenantes+descripción previa de la variante+características moleculares |
| Caso 4 | 47/V | Ejercicio | No | 400 | RyR2(Ile2095Thr) | 2/2 | TVCP: Desencadenante+características moleculares |
| Caso 5 | 66/V | Postoperatorio | No | 416 | RyR2(Gly2367Arg) | 0/0 | TVCP: Características moleculares |
FV: fibrilación ventricular; M: mujer; ms: milisegundos; MSC: muerte súbita cardíaca; SQTC: síndrome de QT corto; TVCP: taquicardia ventricular catecolaminérgica polimórfica; V: varón.

Diagnóstico mediante test genético en caso índice de 23 años con FV y síncopes ante emociones intensas; había presentado ergometría y test de epinefrina positivo, pero presentaba esta variante patogénica en RyR2. El padre de la probando había presentado una MSC y es portador obligado de la mutación.
Los tratamientos instaurados sobre la base de la identificación consistieron en evitar fármacos arritmogénicos y tratamiento de la fiebre en 10 casos de SB, y betabloqueantes en los casos de TVCP y SQTL. Tras un seguimiento medio de 28 meses no se produjo ningún evento en los 19 familiares positivos identificados.
DiscusiónLa etiología y la heredabilidad familiar de la PC de origen no aclarado son aún un reto diagnóstico y clínico, pues es muy escasa la información disponible y las series de pacientes no son amplias como para extraer conclusiones poblacionales. De hecho, las recomendaciones de las últimas guías de actuación en PC de origen incierto de un documento de consenso de la European Heart Rhythm Association (EHRA) se basan en estudios que evaluaron de forma indirecta la afectación familiar en la PC sin cardiopatía10. En general, está recomendado el estudio familiar con test convencionales como ECG y ecocardiograma en todos los casos, y ampliar el estudio con test farmacológicos y de imagen avanzada en caso de hallar anomalías en el probando claramente indicativas del diagnóstico. La realización de test farmacológicos se recomienda con un nivel de evidencia iia para el test de flecainida y iib para el test de epinefrina. Nuestro trabajo, así como el registro FIVI-Gen previamente publicado por nuestro grupo3, aporta por primera vez evidencia sobre la heredabilidad de los trastornos subyacentes en la PC sin cardiopatía aparente y sugiere la necesidad de incluir el estudio familiar, no solo en cascada diagnóstica, sino como una herramienta diagnóstica más.
El trabajo de Kumar et al. de 2013 evaluó la rentabilidad del estudio en familias de sujetos con muerte súbita arrítmica, tanto supervivientes como fallecidos. Ellos encuentran una alta rentabilidad en los casos en que el paciente sobrevive (64%) respecto a aquellos en que no logra sobrevivir a la PC, en los que logran identificar la causa, sobe todo SQTL, en tan solo el 18% de los casos. Este estudio difiere del nuestro fundamentalmente en el criterio de inclusión, es decir, el concepto de PC de etiología incierta, ya que en nuestro caso el ECG con datos de canalopatía suponía la exclusión del estudio y en el suyo no. Nuestra serie, en contraste, evalúa a familias con FV realmente idiopática, en presencia de ECG normal, con un tamaño muestral considerable, demostrando una alta incidencia de casos ocultos entre los familiares que se benefician de un tratamiento preventivo. Es interesante la diferencia que encuentran en la rentabilidad en función de que el sujeto sobreviva o no a la PC: ello se debe a la no disponibilidad del probando en los casos fallecidos para determinar el ECG o test farmacológicos, mostrando tan solo un 18% de diagnóstico familiar en este grupo de ECG no disponible, cifra que se asemeja mucho a nuestros datos, en los que la penetrancia familiar era aproximadamente del 21%.
Un dato muy relevante de nuestro estudio es la demostración de la necesidad de hacer un estudio familiar sistemático, a pesar de no haber encontrado datos fenotípicos indicativos de una patología concreta en el caso índice. Si bien es cierto que es poco frecuente el hallazgo de un ECG anormal cuando el probando no presenta alteraciones, esto ocurrió en 4 de nuestras familias evaluadas (11,4% del total). Trabajos previos no han abordado este tema, sino que el estudio familiar clásicamente se ha orientado «en cascada diagnóstica», una vez alcanzado el diagnóstico en el probando14-20, con una rentabilidad diagnóstica elevada. Esta diferencia es similar a la encontrada por el grupo australiano8 y el razonamiento pasa necesariamente por evaluar tanto a familias de sujetos supervivientes como fallecidos a una PC idiopática de la misma manera, pues se trata del mismo tipo de paciente, y si el estudio familiar es crucial cuando el sujeto ha fallecido, lo debe ser igual cuando el sujeto ha sobrevivido al episodio de FV.
Por último, el impacto clínico de estos casos familiares diagnosticados es evidente: el hecho de identificar una causa hereditaria, con unos factores desencadenantes de arritmias bien identificados, y con un tratamiento farmacológico eficaz en la mayoría de los casos (SQTL y TVCP) como son los betabloqueantes, facilita la prevención de nuevos eventos arrítmicos malignos en la familia. Un trabajo reciente ha evidenciado, no obstante, la baja incidencia de eventos en el seguimiento a largo plazo de familiares de pacientes con muerte súbita arrítmica en ausencia de cardiopatía estructural y diagnóstico definitivo21. Nuestros datos están de acuerdo, con nula presencia de eventos en el seguimiento de los familiares, si bien es cierto que en los casos con diagnóstico identificado se había establecido un tratamiento dirigido en función del fenotipo.
LimitacionesLa limitación fundamental del estudio es, precisamente, la naturaleza subclínica de los trastornos de base causantes de PC sin cardiopatía y su baja expresividad clínica, lo que unido a la sensibilidad limitada de los test farmacológicos y genéticos, según qué patologías, hacen que el número real de familiares afectados sea probablemente superior al observado. Otra limitación es el bajo tamaño muestral y de familiares estudiados; algunos casos rehusaron el estudio y en otros la familia no estaba disponible por haber fallecido los familiares más cercanos, o por adopción. Serían necesarios estudios con mayor tamaño muestral y mayor número de familiares reclutados para confirmar nuestras conclusiones
Otra cuestión relevante son los casos dudosos o limítrofes, en los que el hallazgo en el probando no resultó definitivo: en estos casos el estudio familiar es crucial para verificar la cosegregación del fenotipo y debe realizarse por unidades clínicas con experiencia en el manejo de estas canalopatías y la interpretación cuidadosa de la información genética.
ConclusionesEl estudio familiar es esencial en los casos de PC sin cardiopatía aparente, a pesar de no encontrar alteraciones que dirijan en estudio en el ECG basal o tras test de provocación del caso índice. La prevalencia de familiares con canalopatía oculta fue en torno al 20%, identificados mediante ECG basal, test farmacológicos o test genético dirigido. La identificación correcta de estos familiares permite un tratamiento preventivo, con nula incidencia de eventos arrítmicos en el seguimiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
