



A key process in cell regulation is protein phosphorylation, which is catalyzed by protein kinases and phosphatases. However, phosphoproteomics studies are difficult because of the complexity of protein phosphorylation and the number of phosphorylation sites.
MethodsWe describe an efficient approach analyzing phosphopeptides in single, separated protein by two-dimensional gel electrophoresis. In this method, a titanium oxide (TiO2)-packed NuTip is used as a phosphopeptide trap, together with displacers as lactic acid in the loading buffer to increase the efficiency of the interaction between TiO2 and phosphorylated peptides.
ResultsThe results were obtained from the comparison of mass spectra of proteolytic peptides of proteins with a matrix-assisted laser desorption-ionization-time of flight (MALDI-TOF) instrument.
ConclusionsThis method has been applied to identifying phosphoproteins involved in the symbiosis Rhizobium etli-Phaseolus vulgaris.
Un proceso clave en la regulación celular es la fosforilación de proteínas, que se lleva a cabo por cinasas y fosfatasas. Sin embargo, los estudios de fosfoproteómica son difíciles debido a la complejidad de la fosforilación proteica y el número de sitios de fosforilación.
MétodosEn el presente trabajo se describe una eficiente estrategia metodológica para analizar fosfopéptidos de proteínas separadas mediante electroforesis bidimensional. En este método, una columna con microesferas de dióxido de titanio (TiO2/NuTip) se utilizó para atrapar los fosfopéptidos en la superficie del TiO2 previamente empacado en una punta. El uso de desplazadores en el buffer de carga, como el ácido láctico, mejoró significativamente la selectividad.
ResultadosLos resultados se obtuvieron mediante la comparación de los espectros de masas de péptidos proteolíticos de proteínas analizados utilizando un instrumento de desorción/ionización láser asistida por matriz-tiempo de vuelo (MALDI-TOF).
ConclusionesEste método se ha aplicado para la identificación de fosfoproteínas involucradas en la simbiosis del Rhizobium etli con Phaseolus vulgaris.
Phosphoproteomics is the study of the phosphorylation of proteins that are part of the cell or tissue in a particular state. Phosphorylation is a reversible post-translational modification (PTM), which is key in prokaryotes and eukaryotes biological processes. Protein phosphorylation plays a key role in the regulation of virtually all cellular events. Many crucial biological processes such as cell cycle, cell growth, cell differentiation, and metabolism are strongly controlled by reversible protein phosphorylation, modulating protein activity, stability, interaction, and localization. Phosphorylation is catalyzed by specific protein kinases and phosphatases, and phosphorylation-based signaling is regulated by the antagonizing catalytic activities of protein kinases and protein phosphatases. Activation and deactivation of phosphorylation signaling cascades are relevant in the development of diseases such as cancer1–3.
Large-scale phosphoproteomics studies are difficult because of the complexity of protein phosphorylation and the number of phosphorylation sites. Over 60% of the proteins encoded by some genomes are phosphorylated. It is, therefore, an overwhelming and enormous task to interpret the dynamics of the phosphoproteome4–7. After proteolytic digestions of proteins, a major problem as well is the traceability of any complex modification due to low stoichiometry compared with the unphosphorylated or single phosphorylated peptides. Hence, the development of enrichment methods has given a big boost to the characterization of phosphorylated peptides because it solved the problem of signal suppression by unphosphorylated peptides8. Recently, some strategies relatively sensitive and, in many cases, easier to perform have been developed. For the mass spectrometry(MS)-based strategies, it is common that phosphorylated proteins are enzymatically degraded to peptides, which are subsequently analyzed to detect a mass increment of 80Da per phosphate group. However, none of these MS-based methods can individually provide a complete characterization of a phosphorylated protein of a cellular system in a comprehensive manner and a single experiment9,10.
Enrichment of phosphopeptides can be achieved by two types of methods. The first type includes chromatography-based methods, which include immobilized metal ion affinity chromatography (IMAC) (with Fe3+, Ga3+, Ni2+, and Zr4+ ions), metal oxide affinity chromatography (MOAC) (with TiO2, ZrO2, Al2O3, and Nb2O5), ion-exchange chromatography and hydrophilic interaction liquid chromatography (HILIC)11. Despite the diversity of enrichment strategies, TiO2 is the most commonly used metal oxide for the selective capture of target peptides because it is extremely tolerant toward most buffers and salts used in biochemistry and cell biology. TiO2 has been proven to exhibit a higher affinity and better selectivity for binding phosphopeptides before MS analysis12. The use of selective enhancers is important in this type of methodologies; they improve the binding selectivity of TiO2 toward phosphorylated peptides, thereby enabling phosphorylated peptide characterization from low femtomole level phosphorylated proteins. Chemical reaction-based is the second type of enrichment methods. Phosphopeptides could be covalently conjugated to a polymer support and then released or covalently attached with an affinity tag followed by affinity purification. After enrichment, phosphopeptides are characterized by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF-MS) or submitted to liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) analysis for the identification of phosphopeptides and phosphorylation sites. In the last years, it has been demonstrated that different methods are complementary and can be combined to provide an aggregated data set which is larger than that obtained by a single method. Titanium was first applied as chemoaffinity medium for organophosphates including phosphopeptides and was subsequently made commercially available for proteomics applications13,14. Previously, the inclusion of several additives in the sample loading buffer, such as aromatic hydroxycarboxylic and aliphatic compounds, had been proven to reduce nonspecific binding. These compounds have shown dramatic enrichment improvement during phosphopeptide enrichment with titanium before MS analysis, although their exact physicochemical mechanism is largely unknown15.
Two-dimensional gel electrophoresis (2DE) analysis has commonly been used in global phosphoproteomics studies to separate highly complex protein samples according to their molecular weight (MW) and isoelectric point (pI). Fluorescent stains, such as Pro-Q® Diamond, are commonly used for detection of phosphoproteins16. Hopper et al.17 have shown that Pro-Q® Diamond staining is comparable to 32P labeling regarding sensitivity. In the present investigation, we described a batch mode protocol to identify proteins expressed by Rhizobium etli during symbiosis with Phaseolus vulgaris, using MOAC (TiO2/NuTip) and lactic acid in the loading buffer, which significantly reduced unspecific binding from non-phosphorylated peptides. We also characterized putative phosphoproteins, testing the method with protein candidates separated by 2DE and stained with Pro-Q® Diamond.
2Methods2.1Phosphopeptide enrichment chemicalsPhosphopeptide positive control set from bovine β-casein (monophosphopeptide), DL-lactic acid, trifluoroacetic acid (TFA), dithiothreitol (DTT), and iodoacetamide (IAA) were purchased from Sigma. Acetonitrile HPLC grade was purchased from Fisher Science and water was obtained from a Milli-Q system (Millipore, Bedford, MA). Sequencing grade modified trypsin was obtained from Promega.
2.2Electrophoresis equipment and chemicalsThe Millipore Investigator 2DE system was used (Genomics Solutions, Ann Arbor, MI) for isoelectric focusing (IEF) and sodium dodecyl sulfate (SDS) electrophoresis. Ammonium persulfate, 2-mercaptoethanol (2-ME), bisacrylamide, acrylamide, SDS, and DTT were purchased from BioRad (Richmond, CA). Coomassie brilliant blue G-250 (Fisher Scientific, Fair Lawn, NJ) and Pro-Q stain, urea, and thiourea, from Invitrogen (Carlsbad, CA). TEMED and all other chemicals (analytical grade) for electrophoresis and staining were obtained from Research Organics, Percoll. IGEPAL CA-630, bromophenol blue, Trizma base, Tris hydrochloride, phosphatase inhibitors and phenol were from Sigma (St. Louis, MO). Acetone, acetic acid, methanol, glycerol, and EDTA were from J.T. Baker (Ecatepec, MEX). CHAPS and protease inhibitor cocktail tablet, from Roche (Germany). Ampholytes, NaOH, and phosphoric acid, from Genomic Solutions (Ann Arbor, MI). Unless otherwise indicated, water used for all solutions was 18 mega-ohm purified, filtered through a 0.45μm filter.
2.3Sample preparationNodules were obtained 18 days after inoculation; plants and inoculants were prepared as described previously18. Proteins were extracted using the phenol extraction method with some modifications19,20. In every PBS solutions, phosphatase and protease inhibitors listed before were used during bacteroids separation and phenolic extraction. Proteins were solubilized in the IEF sample solubilization buffer, which is composed by 7 M urea, 2M thiourea, 4% CHAPS, 60mM DTT and 5% v/v carrier ampholytes. Proteins were quantified using Bradford21 assay (Bio-Rad) and stored at -80°C until analyzed.
2.4Protein electrophoresis and visualizationPellets from acetone precipitation were subjected to 2DE, as described in protocols and the 2D Investigator instruction manual (Genomic Solutions, Ann Arbor, MI). pH 4 to 10 ampholytes were used for the first dimension. Preparative 2DE was used to obtain proteins for MS identification (MALDI-TOF-MS or LC-ESI-MS/MS): 1mg of protein was applied using a pH range 4-8. Subsequently, the separation proteins were stained with Pro-Q22 following the manual instructions. After visualization, the stained gel was washed and stained with colloidal Coomassie brilliant blue23. These gels were digitized at 127 X 127μm resolution using a GS-800 Calibrated Densitometer and PD-Quest software (Bio-Rad, Hercules, CA); pI and MW were determined by using a standard 2D SDS-PAGE (Bio-Rad, Hercules, CA).
2.5Mass spectrometry protein identificationProtein spots from Coomassie stained preparative 2D gels were selected and excised manually using a clean scalpel or razor blade and processed. The samples prepared for MALDI-TOF-MS were reduced, alkylated and digested in the gel with trypsin (Promega, Madison, WI) and extracted as previously described20. Before MS analysis, peptides were desalted using a C18 ZipTip (Millipore, Bedford, MA) according to the manufacturer's instructions. Mass spectra were obtained by MALDI-TOF MS.
2.6MALDI-TOF MSMALDI-TOF analysis was carried out with a mass spectrometer (Autoflex, Bruker Daltonics, Billerica, MA) operated in the delayed extraction and reflectron mode. Spectra were externally calibrated using a peptide calibration standard (Bruker Daltonics 206095). All mass spectra were obtained with 200 laser shots. Under delayed extraction conditions, ions were accelerated using 20kV. During the MS analysis, the laser power was adjusted carefully to obtain the highest-quality mass spectra. Peptide mixture was analyzed using a saturated solution of 2,5-dihydroxybenzoic acid (DHB) in 50% acetonitrile/0.1% TFA. Peaks lists of the tryptic peptide masses were generated and searched against the R. etli CFN42, NC_007761.1; pA, NC_007762.1; pB, NC_007763.1; pC, NC_007764.1; pD, NC_004041.2; pE, NC_007765.1; pF, NC_007766.1, and NCBInr databases using the search program Mascot (http://www.matrixscience.com; Matrix Science, Ltd., London, UK).
2.7Nano LC-MS systemA nanoflow HPLC system (Agilent 1100) was used for chromatographic separation of the peptide mixture before MS detection. The injection volume was 8μl and the peptides were eluted at 400 nl/min by an increasing concentration of acetonitrile up to 60% during 32min onto a capillary column Agilent Zorbax 300SB-C18 (3.5μm, 150mm X 75μm). The mobile phases consisted of 0.1% formic acid in 98% aqueous (A) and 0.1% formic acid in 98% acetonitrile (B) solutions.
LC-ESI-MS/MS analysis was performed using Linear 3200 Q Trap (Applied Biosystems), utilizing EMS and NL scan experiments in Q1. The three most abundant ions present in the survey spectrum were automatically mass-selected and fragmented by the collision-induced dissociation. The MS/MS data were searched against the NCBInr protein sequence databases using an in-house Mascot server (Matrix Sciences, London, UK).
2.8Enrichment of phosphopeptides using titanium dioxideThe digestion of α-casein and R. etli proteins obtained from 2D gels was enriched with NuTips filled with TiO2 (1.5mg beads/200μl) purchased from Glygen (Columbia, MD). Monophosphopeptides of β-casein were used as a control of enrichment. Before loading the sample, NuTips were equilibrated with 15μl of solution A (0.1% de TFA, 80% acetonitrile) with 300mg/ml of lactic acid as selectivity enhancer (or displacer). The standard phosphoprotein and extract proteins digested (60μl) were each mixed with 100μl of solution A and 15μl of the sample were loaded in the NuTip for the subsequent interaction of phosphopeptides with TiO2 beads. Then, the NuTip was washed with 15μl of solution A and 15μl of solution B (0.1% TFA, 80% acetonitrile)24. While the phosphopeptides were still bound to the TiO2 beads, 15μl of a solution 0.5% ammonium hydroxide was used for desorbing and elute the phosphopeptides from the samples, and then collected. The eluted fraction was acidified with 0.1% acetic acid and concentrated using C18 ZipTip (Millipore, Bedford, MA) for subsequent MALDI-TOF MS analysis.
3Results and discussion3.1Optimization of enrichment phosphopeptides conditions in TiO2/NuTipThe protocol is based on the selective interaction of water-soluble phosphates with porous TiO2 microspheres via bidentate binding by affinity at the titanium surface. Phosphopeptides are trapped in a NuTip under acidic conditions and desorbed under alkaline conditions. The inclusion of lactic acid in the sample loading buffer was effective to remove acidic non-phosphopeptides, while retained high binding affinity for phosphorylated peptides during enrichment with titanium for MALDI-MS analysis25. The most important purpose of this technique is to increase the relative abundance of phosphorylated peptides minimizing sample complexity. Besides, the chromatography material is directly attached to the inner surface of the tip without polymers or glue, maximizing the surface area in contact with the sample.
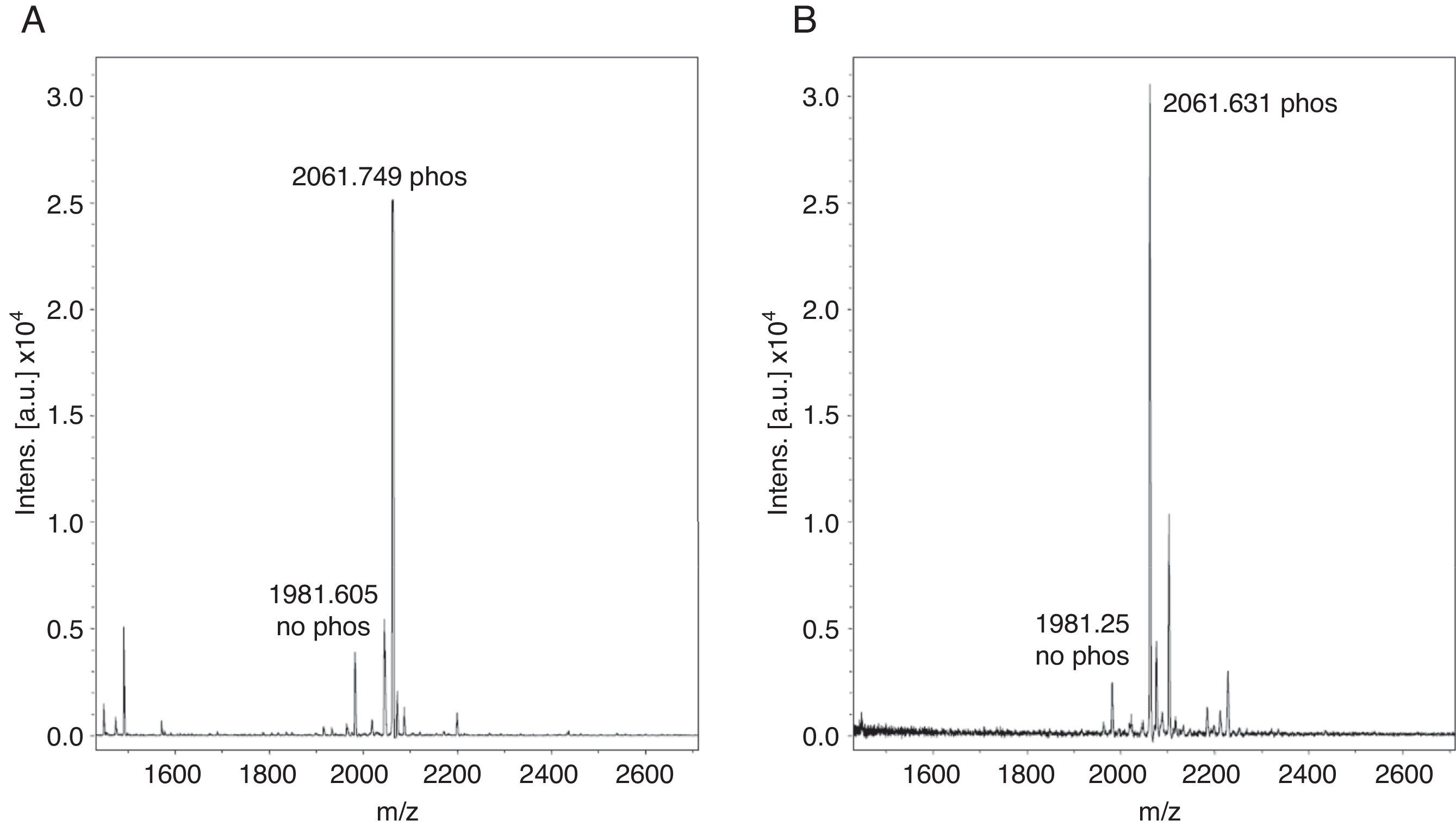
Monophosphopeptide β-casein (13μg) was used as a control in the optimization procedure. A direct analysis showed a lesser relation between non-phosphorylated and phosphorylated peptide FQpSEEQQQTEDELQDK of the standard without TiO2/NuTip (Figure 1A) versus a larger relation obtained after the specific enrichment of monophosphopeptide β-casein in a TiO2/NuTip (Figure 1B).
 MALDI-TOF MS spectra of monophosphopeptide β-casein without enrichment. B) MALDI-TOF MS spectra of monophosphopeptide β-casein enriched with TiO2/NuTip.")
The phosphopeptide selectivity depends on the nature and concentration of displacers; a high concentration improves selectivity but hinders the recovery of phosphopeptides. For this reason, an average concentration of 300mg/ml was chosen for the displacer used in acidic solvent organic loading buffer. Loading solutions typically contain 50-80% acetonitrile concentration to obtain good selectivity, because lower organic concentration results in co-extraction of non-phosphorylated peptides.
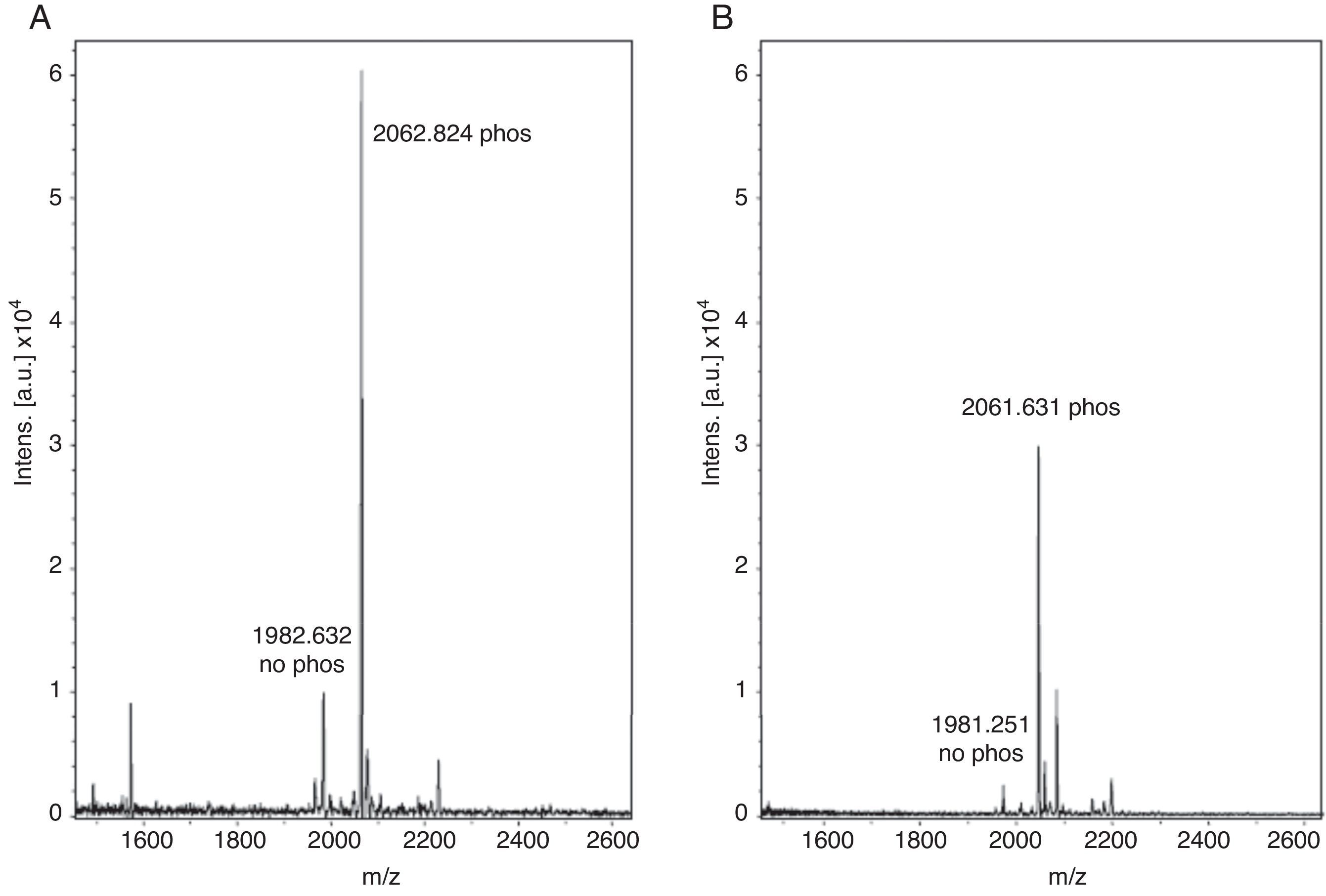
With these benchmarks, the monophosphopeptide β-casein was also enriched with lactic acid as selectivity enhancer in loading buffer (0.1% TFA, 80% acetonitrile, and 300mg/ml lactic acid); 100μl were added to standard and then desorbed phosphorylated peptide from ligand under alkaline conditions. The results evidenced a more intense signal with lactic acid in loading buffer (Figure 2A) versus TiO2/NuTip (Figure 2B) because the aliphatic hydroxycarboxylic acids should bind to metal oxides more weakly than a phosphate group but more strongly than the carboxylic groups of acidic peptides26.
 MALDI-TOF MS spectra of monophosphopeptide β-casein-enriched with NuTip and lactic acid. B) MALDI-TOF MS spectra of monophosphopeptide β-casein-enriched with NuTip but no lactic acid in the loading buffer.")
After enrichment with lactic acid in the loading buffer, there is a competitive elution of the phosphopeptides with acidic peptides and alkaline buffer. Thus, the concentration of phosphopeptides in the matrix of TiO2 is relevant in the process of desorption from the titanium ligand to obtain an intense signal for the phosphorylated peptide. Therefore, the longer the resin is under alkaline conditions, the more phosphopeptides can be released from the ligand in a high concentration to reach saturation, and the greater amount of phosphate groups can be adsorbed to a more negatively charged titanium surface. Some research has demonstrated that the retention of the phosphorylated peptides or proteins depends on the matrix used, pH elution buffer and incubation time with the matrix11.
The enriched sample conformation with lactic acid under alkaline conditions is important for adsorption on titanium because it has a large negatively charged surface, which in some cases cannot be favored to handle a whole. Electrostatic interactions can affect certain compounds but not necessarily in all cases. The conformation of the species formed between the treated sample and the enhancer is more important to be completely adsorbed over the entire surface. Also, a highly alkaline elution buffer system favors a greater more amount of phosphorylated peptides because it helps to desorb peptides highly retained in the matrix27.
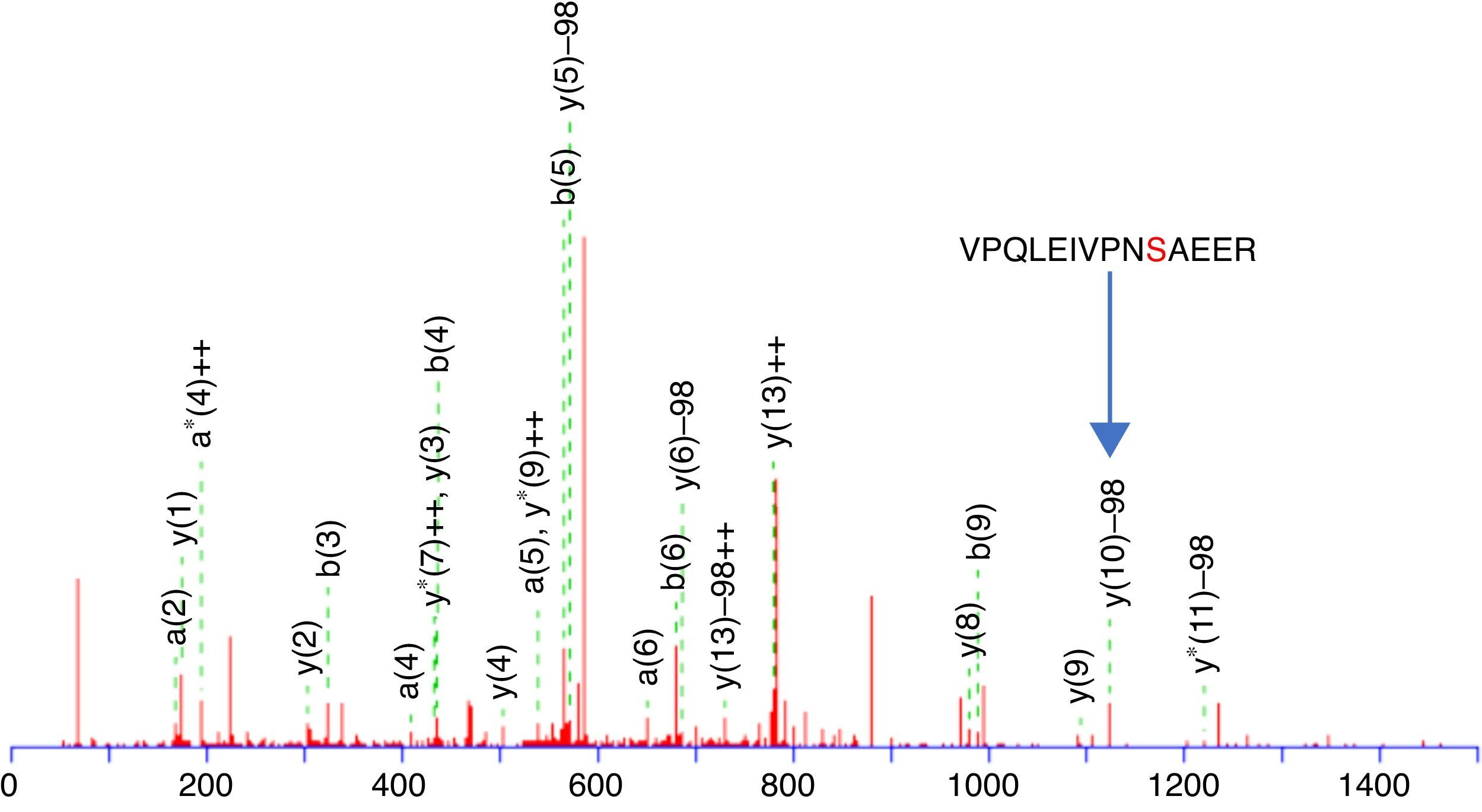
This enrichment proposal was applied successively for the detection and identification of phosphopeptides from tryptic digestion of α-casein standard. The enrichment of α-casein could also be checked on a linear ion trap nano-HPLC-IT-MS with Neutral Loss Scan of 98, 49 or 32.7 (H3PO4 for the +1, +2 and +3 charged ions), where the phosphorylated peptide K.YKVPQLEIVPNSAEER.L with a +2 charge ion mass of 830.8400 and a score of 44 was determined (Figure 3). The injection of the same standard digested, without enriching, was also subjected to the same experiment of Neutral Loss resulting in the identification of α-casein with the same peptide and a score of 11. These results demonstrate the efficiency of enrichment with titanium. The enriched sample score was higher, and the identification of a phosphorylated peptide more clear.
3.2Enrichment of phosphopeptides from proteins obtained from two-dimensional gel electrophoresis
Once the conditions were established with TiO2/NuTip enrichment and lactic acid as the enhancer with a standard, we aimed to demonstrate that that combination was capable of effective enrichment of phosphopeptides from a real sample; a tryptic digest of phosphoprotein candidates from 2D gels detected using specific fluorescence stains (Pro-Q Diamond) was used. They were randomly selected with low and high stoichiometry, also considered covered from different MW and pI for enrichment (Figure 4).

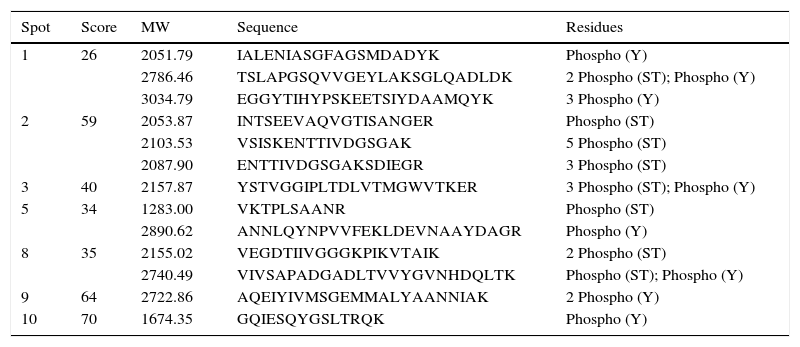
The proteins were cut and digested in the gel to obtain tryptic peptides and could be enriched with the best optimization conditions with TiO2/NuTip. The proteins were identified as WP_011427135 aconitate hydratase (spot 1, Mr 97180, pI 5.48), WP_011424178 heat shock chaperonin (60 kDa) (spot 2, Mr 57682, pI 5.03), WP_016736374 malate dehydrogenase (spot 3, Mr 33850, pI 5.53), WP_011426970 peptidyl prolyl cis-trans isomerase (spot 4, Mr 32231, pI 4.84), WP_020921184 general L-amino acid ABC transporter, substrate-binding (spot 5, Mr 37697, pI 5.44), WP_011053300 hypothetical protein (spot 6, Mr 23755, pI 5.83), WP_004678527 electron transfer flavoprotein alpha subunit (spot 7, Mr 39842. pI 6.72), WP_011426719 glyceraldehyde 3-phosphate dehydrogenase (spot 8, Mr 36016, pI 6.87), WP_040140722 nitrogenase iron protein (spot 9, Mr 32231, pI 4.84), and WP_011425130 trigger factor protein (spot 10, Mr 54412, pI 4.83). The enrichment was developed with lactic acid in loading buffer. Table 1 shows the phosphopeptides sequences with gains of 80Da.
TiO2 enrichment of phosphopeptides from tryptic digestion of proteins expressed by R. etli in symbiosis with P. vulgaris.
| Spot | Score | MW | Sequence | Residues |
|---|---|---|---|---|
| 1 | 26 | 2051.79 | IALENIASGFAGSMDADYK | Phospho (Y) |
| 2786.46 | TSLAPGSQVVGEYLAKSGLQADLDK | 2 Phospho (ST); Phospho (Y) | ||
| 3034.79 | EGGYTIHYPSKEETSIYDAAMQYK | 3 Phospho (Y) | ||
| 2 | 59 | 2053.87 | INTSEEVAQVGTISANGER | Phospho (ST) |
| 2103.53 | VSISKENTTIVDGSGAK | 5 Phospho (ST) | ||
| 2087.90 | ENTTIVDGSGAKSDIEGR | 3 Phospho (ST) | ||
| 3 | 40 | 2157.87 | YSTVGGIPLTDLVTMGWVTKER | 3 Phospho (ST); Phospho (Y) |
| 5 | 34 | 1283.00 | VKTPLSAANR | Phospho (ST) |
| 2890.62 | ANNLQYNPVVFEKLDEVNAAYDAGR | Phospho (Y) | ||
| 8 | 35 | 2155.02 | VEGDTIIVGGGKPIKVTAIK | 2 Phospho (ST) |
| 2740.49 | VIVSAPADGADLTVVYGVNHDQLTK | Phospho (ST); Phospho (Y) | ||
| 9 | 64 | 2722.86 | AQEIYIVMSGEMMALYAANNIAK | 2 Phospho (Y) |
| 10 | 70 | 1674.35 | GQIESQYGSLTRQK | Phospho (Y) |
As mentioned previously, the enrichment of phosphoprotein candidates was developed using lactic acid as an enhancer, corroborating the presence of phosphorylated peptides. For low and higher MW proteins, lactic acid has shown an evidence of 80Da gains of some peptides, confirming protein phosphorylation. This enhancer helped to detect peptides with more than one phosphorylated site (Table 1) and with aspartic acid phosphorylated residues (reported in prokaryotes organisms)28. Also, non-phosphorylated peptides were co-extracted (data not shown).
Two other proteins were processed as controls and identified, a peptidyl-prolyl cis-trans isomerase and a hypothetical protein, which were not stained with the ProQ. The phosphorylated peptides after the enrichment with NuTip were neither obtained (data not shown).
Also, it was not possible to detect another two proteins (spots 6 and 7) as phosphoproteins, one electron-transfer flavoprotein and one hypothetical protein, which were originally detected with Pro-Q Diamond, suggesting a deficient specificity of the Pro-Q Diamond staining. Unlike IMAC, which gets more non-specific interactions and increases the possibility of false positives, the coverage of most proteins analyzed showed selectivity and specificity of the enrichment procedure by MOAC. In addition, it represents a better alternative to the esterification of carboxylic acids of peptides because is incomplete and its solubility decreases after esterification, leading to greater loss of sample.
When lactic acid was introduced into the loading buffer, non-phosphopeptides were significantly inhibited, and the specific binding was greatly improved. Under the optimal conditions, the TiO2–lactic acid showed good results for enriching phosphopeptides at different concentration levels. Besides, the enrichment methodology—applied in the phosphorylated proteins identification detected in 2D gels stained candidates with the Pro-Q dye—was successful. This enrichment method allowed detecting peptides phosphorylated in more than one residue.
An important number of non-phosphorylated peptides were observed. These peptides contained aspartic and glutamic acid residues accompanied with hydrophobic amino acids such as valine, leucine, and phenylalanine (data not shown). The interaction between the titanium acid peptides has already been discussed in previous works27.
Phosphopeptides with basic residues also have low affinity with TiO2, which affects its enrichment in a negative way. These results are to be expected since the nonspecific binding cannot be completely suppressed due to the high range of concentration, which is a characteristic of peptides present in the protein digestion. Furthermore, the incomplete union between the phosphorylated peptides and the titanium support should also be considered in the cases for the peptidyl-prolyl cis-trans isomerase, the hypothetical protein, and electron-transfer flavoprotein alpha subunit, in which there were not enriched phosphorylated peptides (spots 4, 6, 7). As mentioned previously in the results section, another possibility could be the deficient specificity of Pro-Q Diamond staining. Further experiments are needed to clarify this issue.
Despite the nonspecific binding of non-phosphorylated peptides and the phosphopeptides loss due to the additional separation process, it was possible to obtain important information about the post-transcriptional modifications with this methodology, particularly in this case of some R. etli proteins and its possible role in the symbiosis with P. vulgaris.
Using an instrument device like the MALDI-TOF, rapid, efficient results could be observed before determining the exact phosphorylation site in a particular sequence with more specialized techniques, such as Neutral Loss and Precursor Ion Scan, saving analysis time.
Ethical responsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingThis work was supported by combined grants from National Council of Science and Technology CONACyT-Mexico, Grant 220790, and from PAPIIT-DGAPA UNAM, Grant IN213216.
Conflict of interestThe authors declare no conflicts of interest of any nature.
We thank Oliver Castillo, J. L. Zitlalpopoca, and Hadau Sánchez for plant experiments and greenhouse support.