


La participación del canal de Ca2+/receptor de rianodina en el acoplamiento excitación-contracción cardiaco se conoce desde finales de los años ochenta, cuando en varios trabajos trascendentales se comunicó por primera vez su purificación y se encontró que correspondía a las estructuras conocidas como «pies» localizadas en las cisternas terminales del retículo sarcoplásmico. Adicionalmente a su papel como canal responsable del aumento global y transitorio de Ca2+ que activa a la maquinaria contráctil durante el ciclo cardiaco, el receptor de rianodina también libera Ca2+ durante la fase de relajación, dando lugar a la fuga de Ca2+ en la diástole que en condiciones fisiológicas regula el nivel de Ca2+ luminal, pero cuando se encuentra alterada participa en la generación de arritmias adquiridas o hereditarias. Recientemente, el esfuerzo de diversos grupos de investigación se ha enfocado en el desarrollo de herramientas farmacológicas para controlar la fuga diastólica de Ca2+ que se presenta alterada en algunas enfermedades cardiacas. En esta revisión nos enfocamos en describir la participación del receptor de rianodina cardiaco en la fuga diastólica de Ca2+ así como los diversos enfoques terapéuticos que se han implementado para controlar su actividad exacerbada en la diástole.
The participation of the ionic Ca2+ release channel/ryanodine receptor in cardiac excitation-contraction coupling is well known since the late ’80s, when various seminal papers communicated its purification for the first time and its identity with the “foot” structures located at the terminal cisternae of the sarcoplasmic reticulum. In addition to its main role as the Ca2+ channel responsible for the transient Ca2+ increase that activates the contractile machinery of the cardiomyocytes, the ryanodine receptor releases Ca2+ during the relaxation phase of the cardiac cycle, giving rise to a diastolic Ca2+ leak. In normal physiological conditions, diastolic Ca2+ leak regulates the proper level of luminal Ca2+, but in pathological conditions it participates in the generation of both, acquired and hereditary arrhythmias. Very recently, several groups have focused their efforts into the development of pharmacological tools to control the altered diastolic Ca2+ leak via ryanodine receptors. In this review, we focus our interest on describing the participation of cardiac ryanodine receptor in the diastolic Ca2+ leak under physiological or pathological conditions and also on the therapeutic approaches to control its undesired exacerbated activity during diastole.
El ion calcio (Ca2+) es el mensajero más importante en el músculo cardiaco, ya que participa activamente en diversos procesos celulares tales como la expresión génica, la diferenciación y la apoptosis, además de ser componente indispensable del proceso de contracción, tal como lo mostró Sidney Ringer1. Las fluctuaciones periódicas en la concentración intracelular de Ca2+ ([Ca2+]i) de los cardiomiocitos determinan en gran medida la magnitud y la duración de la fuerza contráctil. La serie de eventos exquisitamente coordinados mediante los cuales el potencial de acción cardiaco genera un aumento global y transitorio en la [Ca2+]i que alcanza niveles de entre 0.6 a 3μM en la sístole y que permite la contracción subsecuente de los miocitos cardiacos se engloban en lo que conocemos como acoplamiento excitación-contracción (AEC), visualizado inicialmente por A. J. Brady2 y descrito con mucha más precisión por Fabiato3,4.
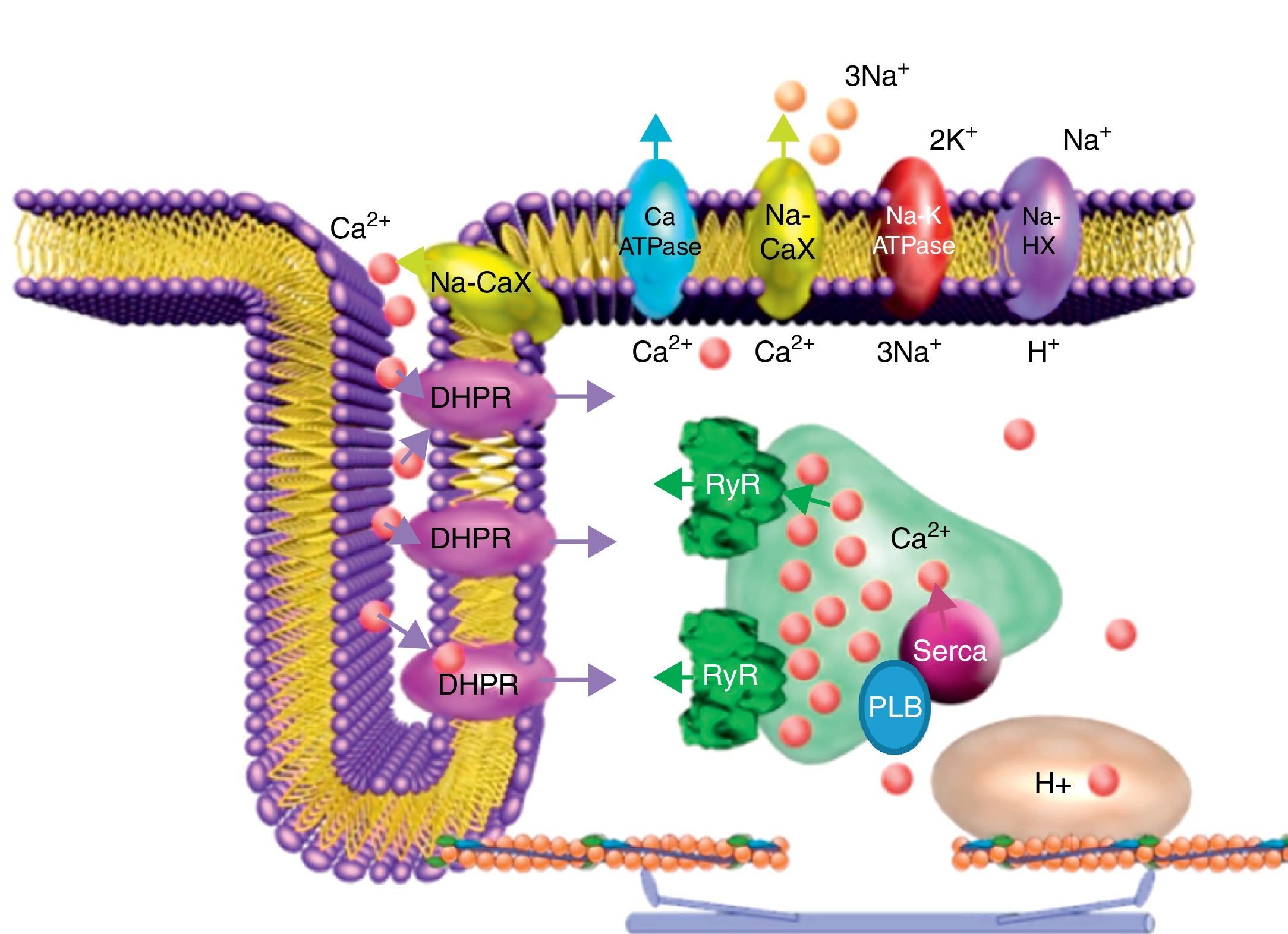
Participación del receptor de rianodina en el acoplamiento excitación-contracciónDurante el AEC cardiaco la activación en el cardiomiocito de los canales de Na+ dependientes de voltaje (INa) activa a su vez los canales de Ca2+ dependientes de voltaje tipoL o receptores a dihidropiridinas (DHPR, por sus siglas en inglés) localizados en una región muy especializada de la membrana plasmática conocida como túbuloT (fig. 1), lo que genera una corriente entrante de Ca2+(ICa) que por sí sola es insuficiente para inducir la activación de la maquinaria contráctil pero es necesaria para activar al receptor de rianodina (RyR), que en realidad es un canal liberador de Ca2+ del retículo sarcoplásmico de unión (RSU). Este proceso se conoce como liberación de Ca2+inducida por Ca2+ (CICR), y fue descrito por Fabiato4 para el músculo cardiaco, pero definido inicialmente por Endo et al.5 en el músculo esquelético.
 activa a los receptores de rianodina (RyR) localizados en el RSU, lo que induce la liberación de cantidades mayores de Ca2+ de los reservorios intracelulares aumentando rápidamente la concentración citoplasmática de Ca2+. Esto permite que se active la maquinaria contráctil. Durante la relajación se promueve el cerrado del RyR y la remoción de Ca2+ se lleva a cabo principalmente por la bomba ATPasa de Ca2+ del RS (SERCA), cuya función está regulada por fosfolamban (PLB) y que se encarga de recuperar los reservorios para el siguiente ciclo del AEC; el intercambiador Na+/Ca2+ (Na-CaX) remueve parte del Ca2+ hacia al exterior de la célula, y de forma minoritaria la bomba de Ca2+ de la membrana plasmática (Ca-ATPase). Las flechas indican la dirección de los flujos de Ca2+ vía los canales iónicos (DHPR, RyR), intercambiadores (Na-CaX) y bombas (SERCA, Ca-ATPase). Na-K ATPase, bomba ATPasa de Na+ y K+; Na-HX, intercambiador Na+/H+.")
Participación del receptor de rianodina en el acople excitación-contracción cardiaco. El influjo de Ca2+ vía los canales de Ca2+ dependientes de voltaje tipoL o receptores a dihidropiridinas (DHPR, localizados principalmente en los túbulosT) activa a los receptores de rianodina (RyR) localizados en el RSU, lo que induce la liberación de cantidades mayores de Ca2+ de los reservorios intracelulares aumentando rápidamente la concentración citoplasmática de Ca2+. Esto permite que se active la maquinaria contráctil. Durante la relajación se promueve el cerrado del RyR y la remoción de Ca2+ se lleva a cabo principalmente por la bomba ATPasa de Ca2+ del RS (SERCA), cuya función está regulada por fosfolamban (PLB) y que se encarga de recuperar los reservorios para el siguiente ciclo del AEC; el intercambiador Na+/Ca2+ (Na-CaX) remueve parte del Ca2+ hacia al exterior de la célula, y de forma minoritaria la bomba de Ca2+ de la membrana plasmática (Ca-ATPase). Las flechas indican la dirección de los flujos de Ca2+ vía los canales iónicos (DHPR, RyR), intercambiadores (Na-CaX) y bombas (SERCA, Ca-ATPase). Na-K ATPase, bomba ATPasa de Na+ y K+; Na-HX, intercambiador Na+/H+.
El Ca2+ liberado vía el RyR se une a la troponinaC de los miofilamentos, lo que genera un cambio conformacional en el complejo de la troponina que libera el sitio de interacción miosina-actina y con gasto de ATP se activa la maquinaria contráctil. Para que la relajación ocurra durante el ciclo cardiaco y la [Ca2+]i disminuya a su nivel diastólico (∼100nM), se requiere re-secuestrar el Ca2+ liberado por el RyR mediante la activación de la bomba ATPasa de Ca2+ del retículo sarco/endoplásmico (SERCA), mientras que el Ca2+ que entró como ICa debe ser extruido por el intercambiador Na+/Ca2+ de la membrana plasmática. Ambas actividades combinadas remueven hasta el 98% del Ca2+ citoplasmático. La contribución de la bomba ATPasa de la membrana plasmática y del uniportador de Ca2+ mitocondrial en la remoción del Ca2+ citoplasmático es minoritaria (solo ∼1% del Ca2+ total)6.
Siendo el Ca2+ el agonista fisiológico del RyR, la salida masiva de Ca2+de los reservorios intracelulares durante el proceso de CICR implicaría la existencia de un mecanismo de retroalimentación positiva que mantendría activado al RyR de forma permanente (presencia de Ca2+=activación del RyR). Sin embargo, la fase de diástole del ciclo cardiaco depende de la regulación precisa del RyR y de su inactivación aun en presencia de niveles elevados de Ca2+ en el citoplasma para permitir la recuperación de la [Ca2+]i al nivel nanomolar (∼100nM). Las alteraciones en el manejo de Ca2+ sistólico7,8 y diastólico9,10 del cardiomiocito pueden participar en la disfunción contráctil y en la generación de algunas arritmias. De ahí la importancia de estudiar los mecanismos que participan en la regulación del RyR.
Debido a que la activación del RyR cardiaco es en esencia autorregenerativa11, esta requiere de uno o varios mecanismos de inactivación que dependen en gran medida de la presencia de iones y metabolitos citoplasmáticos12,13 (como Ca2+, Mg2+ y ATP), de proteínas reguladoras (calmodulina14, sorcina15, FKBP12.616,17, S100A, junctina, triadina y calsecuestrina18,19, entre muchas otras), de modificaciones postraduccionales (como fosforilación16,20-22, oxidorreducción23, S-nitrosilación24 y carbonilación25), así como de la distribución y localización espacial de los RyR en el cardiomiocito26,27. Los mecanismos moleculares de regulación endógena del RyR cardiaco han sido sujeto de numerosas y excelentes revisiones28-33.
Dentro de los procesos que pueden participar en la terminación de la liberación de Ca2+ vía el RyR se han postulado a: 1)la inactivación/adaptación del RyR dependiente de Ca2+, que involucra al Ca2+ tanto de lado del citoplasma como de la región luminal del RS34 y a proteínas cuya actividad sobre el RyR depende de Ca2+, como sorcina35,36, calmodulina14 y calsecuestrina18,19; 2)el decaimiento espontáneo de la actividad de los RyR debido al cerrado estocástico del canal (stochastic attrition)37; 3)el agotamiento de las reservas de Ca2+ luminal del RS que induce la desactivación del RyR11,19, y por último 4)el «decaimiento inducido» de la actividad de los RyR, una propiedad intrínseca a la microdistribución y activación estocástica de los RyR que permite el aumento y la disolución casi instantánea de los gradientes de Ca2+ nanoscópicos dentro de una región local27. Esta última propuesta viene a formar parte de las teorías de control local de la liberación de Ca2+ durante el AEC cardiaco38 que han ayudado a explicar cómo un fenómeno en esencia autorregenerativo como el de CICR puede ser limitado espacial y temporalmente para permitir una liberación gradual del Ca2+ del RS durante la sístole. Actualmente no se ha encontrado un mecanismo de terminación de la liberación de Ca2+ vía el RyR lo suficientemente eficiente que permita explicar el cerrado preciso del canal durante el AEC cardiaco11,27. Aunque es posible que varios de los mecanismos mencionados funcionen en unísono, se ha propuesto que la inactivación de los RyR dependiente de Ca2+ luminal juega un papel preponderante como mecanismo para detener la naturaleza autorregenerativa inherente al fenómeno de CICR34.
Distribución espacial y regulación in situ de los receptores de rianodina cardiacosAlgunas de las teorías actuales para explicar la terminación de la salida de Ca+ de los reservorios intracelulares —como la del «decaimiento inducido» de la actividad del RyR27— requieren como premisa una distribución precisa de estos canales iónicos; de ahí la importancia de estudiar la microarquitectura de las diadas, o regiones de acoplamiento entre los RyR que se encuentran en el RSU y los DHPR del túbuloT (fig. 1).
El RyR cardiaco es el canal iónico más grande conocido hasta el momento. El canal funcional es un tetrámero, cada monómero está constituido por 4,967 residuos de aminoácidos (RyR2-human entry: Q92736, http://www.uniprot.org) y aparece como una banda prominente, con un peso molecular (Mr) en geles desnaturalizantes (SDS-PAGE) de 340,000 y en ocasiones acompañada de una banda de menor tamaño (Mr de 300,000)39, aunque esto último varía dependiendo de las condiciones en que se preparan las vesículas enriquecidas de RS que contienen mayoritariamente al RyR, o bien de las condiciones del SDS-PAGE. Aunque es un solo gen el que codifica a la isoforma cardiaca del RyR (RYR2, localizado en el cromosoma1 del humano, locus 1q43)40, este cuenta con 105 exones que por procesamiento alternativo del mRNA pueden generar por lo menos 2variantes del RyR2. Estas variantes tienen capacidades diferentes en la regulación del Ca2+ intracelular y de la apoptosis41. Gracias al alcaloide rianodina (proveniente de la planta Ryania speciosa) se pudo purificar39,42 y determinar la localización subcelular del receptor, el cual se encuentra principalmente en la región del RSU (o cisternas terminales), precisamente donde se encontraban las estructuras conocidas como «pies», que por su tamaño relativamente grande (210-220Å por lado) podían ser visualizadas por microscopia electrónica, lo que permitió determinar la identidad molecular de estas estructuras39,42-44.
Datos recientes de microscopia óptica de alta resolución muestran que los RyRs cardiacos del RSU se encuentran formando grupos («racimos» o clústers) dentro de las unidades de liberación de Ca2+ (o CRU, de calcium release units)26,45. Los RyR agrupados presentan un arreglo cuasi-cristalino en la región del RSU y se distribuyen de forma regular formando filas dobles con un ancho aproximado de 700nm con muy pocos RyR en las regiones intermedias. Dentro de la CRU hay una distancia de 10 a 15nm entre los RyR y los DHPR, lo que permite su interacción cercana26. Interesantemente, los clústers de RyR varían en forma y tamaño, existiendo en arreglos de entre 14 hasta 22RyR dentro de cada uno. Un pequeño porcentaje de clústers cuenta con 100 RyR (es posible determinar el número de RyR asumiendo un área de 900nm2 por canal para dividir el área de cada grupo). Esta organización sigue una distribución que se ajusta a un decaimiento monoexponencial, sugiriendo que la formación del clústers es un proceso estocástico y no altamente regulado, como se creía anteriormente26. La distancia entre los RyR dentro del clúster ha sido calculada en 30nm a partir del centro del poro, sugiriendo que se encuentran interactuando físicamente, mientras que entre grupos adyacentes de RyR la distancia de borde a borde es de entre 50 a 100nm, lo que sugiere que la comunicación intragrupo está más favorecida26. Por otra parte, en 2006 Sobie et al.46 postularon la existencia de RyR que se encuentran fuera de los clústers, conocidos también como «RyRs rebeldes» (rogue RyRs), cuya presencia ha sido confirmada con la microscopia óptica de alta resolución26 en zonas del RS clásicamente conocidas como RS corbular44, y que son regiones del RSU que contienen RyR pero que no están asociadas al túbuloT o a la membrana plasmática.
Dentro de las CRU, los RyR y los canales de Ca2+ dependientes de voltaje se distribuyen en regiones de ∼100nm de diámetro. La organización de los RyR y los canales de Ca2+ dependientes de voltaje en las CRU tiene implicaciones biofísicas importantes en cuanto a su funcionalidad en la señalización de Ca2+, y en principio un solo canal de Ca2+ dependiente de voltaje puede activar hasta 5RyR47-49. Recientemente se propuso un modelo donde cada CRU contiene un clúster principal de RyR, que se puede dividir en subdominios funcionales, rodeado por RyR rebeldes, con acceso compartido a un mismo reservorio de Ca2+ luminal; este modelo puede ayudar a explicar la existencia de las diferentes modalidades de liberación espontánea de Ca2+ del RS en condiciones pasivas, como las chispas de Ca2+ y las ondas de Ca2+, que forman parte de lo que conocemos como «fuga de Ca2+ diastólica»46.
Participación del receptor de rianodina en la fuga de Ca2+ diastólicaBajo condiciones fisiológicas, la activación del RyR durante el AEC cardiaco genera un incremento del Ca2+, homogéneo y transitorio, en el citoplasma del cardiomiocito6,50. Sin embargo, en la diástole, al contrario de lo que se pensaba, el RyR no permanece completamente cerrado, sino que puede participar en la liberación de Ca2+ del RS conocida como fuga de Ca2+ diastólica46, la cual está presente en condiciones fisiológicas normales y sufre alteraciones que promueven actividad arrítmica en ciertas afecciones cardiacas.
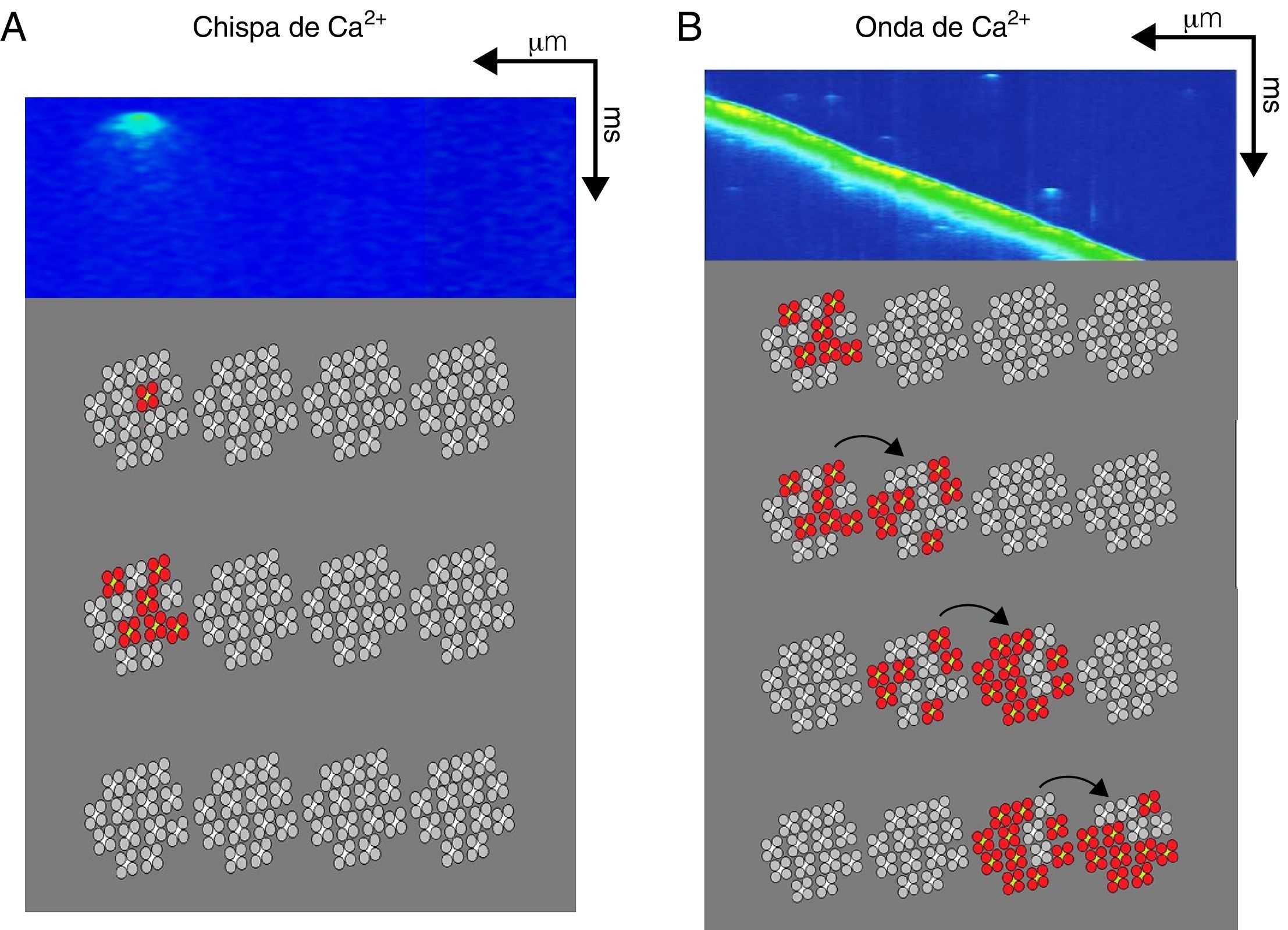
La fuga de Ca2+ diastólica se puede clasificar en: 1)la mediada por la activación de los RyR dentro de una CRU y que se manifiesta en forma de chispa de Ca2+ (fig. 2); la corriente de Ca2+ de un solo canal de RyR oscila entre 0.35 y 0.6pA (con una [Ca2+]RS=1mM), y esto implica que para la generación de un evento elemental de liberación de Ca2+ dentro de una CRU (chispa de Ca2+ o Ca2+spark) es necesaria la apertura sincronizada de por lo menos 6RyR, aunque el número real es difícil de calcular47-49; 2)la que permite el reclutamiento secuencial de CRU adyacentes generando ondas de Ca2+ autopropagadas (fig. 2); si las ondas de Ca2+ activan una corriente transitoria entrante vía el intercambiador Na+/Ca2+(Iti), pueden alcanzan el umbral de disparo de un potencial de acción generando actividad automática50,51. La actividad automática participa en la generación de arritmias por medio de posdespolarizaciones, que pueden ocurrir dentro (posdespolarizaciones tempranas, o early after depolarizations [EAD]) o después de completar la fase de repolarización del potencial de acción (posdespolarizaciones tardías o delayed after depolarizations [DAD]), generando contracciones fuera de ritmo (poscontracciones o after contractions); 3)la que no se puede detectar experimentalmente con los microscopios actuales y que está mediada por los RyR rebeldes, conocida como fuga invisible, y por último 4)la mediada por otros canales de la membrana del RS diferentes al RyR (p.ej., el receptor de 1,4,5-trifosfato de inositol) y que son responsables de la fuga de Ca2+ diastólica aun en presencia de altas concentraciones de rojo de rutenio, un inhibidor de los RyR34,52.
 y una onda de Ca2+espontánea (B). En la parte inferior de cada imagen se esquematizan 4unidades de clústers de RyR con 13canales iónicos en promedio (tetrámeros en gris claro y centro blanco). La generación de una chispa de Ca2+ (A) requiere la activación cuasisimultánea de al menos 6RyR (tetrámeros en rojo) dentro de un clúster que generan una señal local de Ca2+ que no propaga a los clústers adyacentes. Por otra parte, las ondas de Ca2+ espontáneas (B) implican la propagación de la señal de Ca2+ a clústers de RyR adyacentes mediante el mecanismo de activación-difusión-activación que pueden generar contracciones fuera del ritmo. Por último, la fuga de Ca2+ que no se puede observar por microscopia confocal (fuga invisible) involucra la apertura de un número reducido (tal vez solo uno) de RyR que pueden localizarse principalmente fuera de los clústers.")
Participación del receptor de rianodina en la fuga de Ca2+ en la diástole. Imágenes bidimensionales obtenidas por microscopia confocal en donde se observa una chispa de Ca2+ (A) y una onda de Ca2+espontánea (B). En la parte inferior de cada imagen se esquematizan 4unidades de clústers de RyR con 13canales iónicos en promedio (tetrámeros en gris claro y centro blanco). La generación de una chispa de Ca2+ (A) requiere la activación cuasisimultánea de al menos 6RyR (tetrámeros en rojo) dentro de un clúster que generan una señal local de Ca2+ que no propaga a los clústers adyacentes. Por otra parte, las ondas de Ca2+ espontáneas (B) implican la propagación de la señal de Ca2+ a clústers de RyR adyacentes mediante el mecanismo de activación-difusión-activación que pueden generar contracciones fuera del ritmo. Por último, la fuga de Ca2+ que no se puede observar por microscopia confocal (fuga invisible) involucra la apertura de un número reducido (tal vez solo uno) de RyR que pueden localizarse principalmente fuera de los clústers.
La fuga de Ca2+ diastólica que no involucra a los RyR es insensible a las variaciones en la concentración de Ca2+ luminal ([Ca2+]RS), mientras que la fuga de Ca2+ mediada por los RyR (localizados dentro o fuera de los clústers) es regulada por la [Ca2+]RS, por la [Ca2+]i pero no necesariamente por el nivel de fosforilación de los RyR52,53. En condiciones fisiológicas normales, cuando la [Ca2+]RS es relativamente baja (<400μM), la cantidad de Ca2+ liberada por un RyR es insuficiente para generar una chispa, dando lugar a la fuga invisible. Al incrementarse la [Ca2+]RS (>600μM), el flujo de salida de Ca2+ de un RyR es suficiente para que el Ca2+ alcance los sitios de activación por Ca2+ de RyR vecinos, propagándose la activación dentro del clúster, y por tanto la fuga de Ca2+ se presenta en forma de chispas de Ca2+ (fig. 2). Finalmente, en situaciones donde la [Ca2+]RS se incrementa a niveles que alcanzan la sobrecarga del RS, la fuga de Ca2+ mediada por uno o varios clústers puede reclutar clústers adyacentes promoviendo la aparición de ondas de Ca2+52.
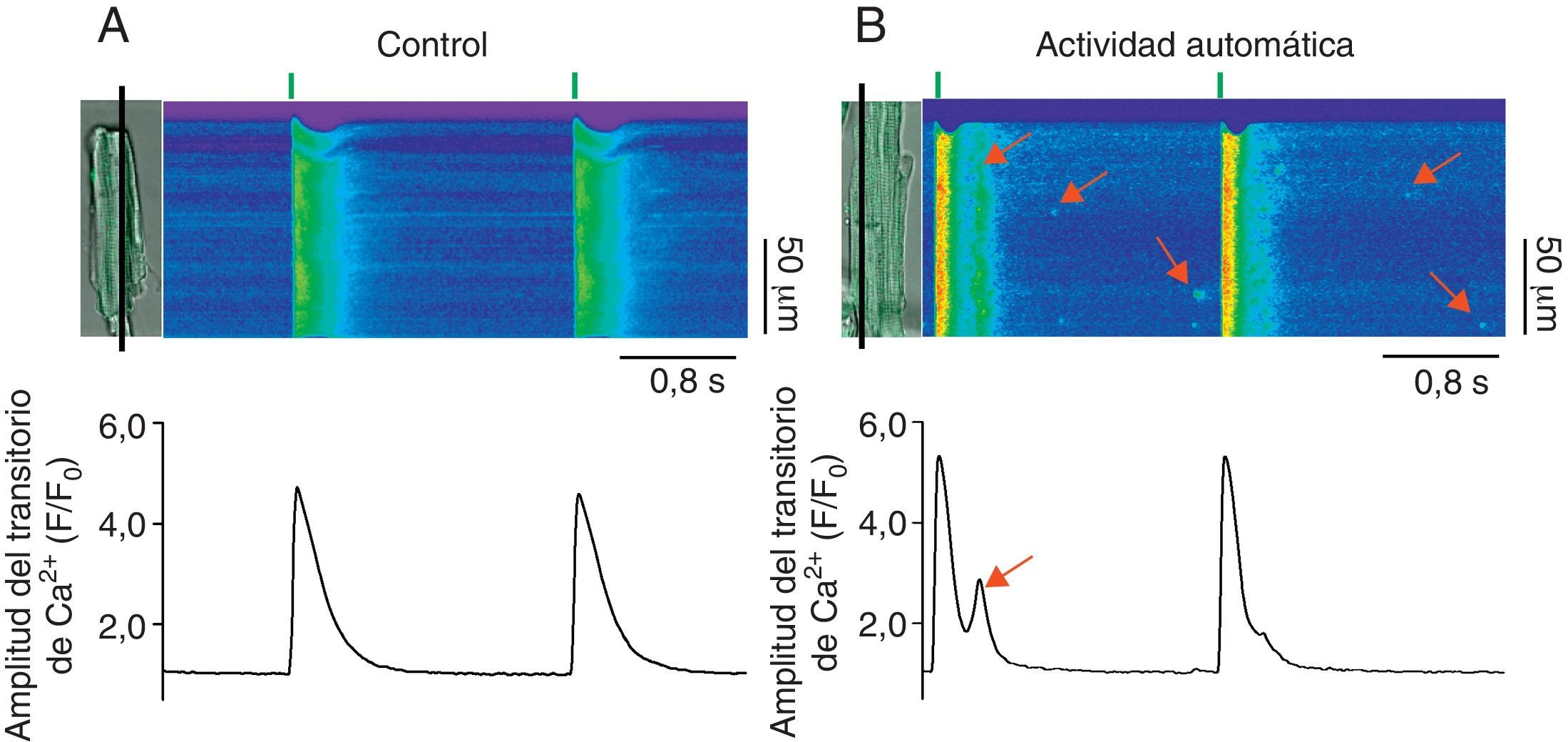
En ciertos procesos patológicos donde la fuga de Ca2+ diastólica está alterada, la actividad anormal del RyR genera ondas de Ca2+ que pueden activar al intercambiador Na+/Ca2+ de la membrana plasmática. Debido a que el intercambiador Na+/Ca2+ es electrogénico, genera una corriente entrante que si alcanza el umbral de activación de los DHPR promueve DAD, trastorna la función contráctil (al generar extrasístoles) y se manifiesta en arritmias (fig. 3)34,52–54. Así, la actividad anormal del RyR en la diástole promueve la arritmogénesis vía la generación de ondas de Ca2+ autopropagadas, la activación del intercambiador Na+/Ca2+ y la aparición de DAD55–58. Es importante mencionar que la fuga de Ca2+ diastólica en forma de chispas que no se propagan a ondas o la mediada por RyR no acoplados (fuga invisible) difícilmente podría promover DAD y, por lo tanto, actividad arritmogénica. El estudio de las propiedades espaciotemporales de las chipas de Ca2+ que se producen de forma espontánea en cardiomiocitos que se encuentran en condiciones en reposo (equivalentes a la diástole) permite conocer cómo los RyR participan en la generación de DAD.
 en cardiomiocitos control (A, control) y con fuga de Ca2+ anormal durante la diástole (B, actividad automática). Las células cargadas con el indicador Fluo-3 se escanearon en la modalidad de line-scan en el microscopio confocal (línea negra), lo que permite generar las imágenes bidimensionales donde el ejeX representa el tiempo en segundos y el ejeY representa la longitud de la célula en micrómetros. La amplitud del transitorio de Ca2+ intracelular (F/F0) producido por el estímulo eléctrico se muestra debajo de cada imagen. Las flechas rojas en las imágenes bidimensionales indican la fuga de Ca2+ durante la diástole en forma de chispas de Ca2+ y ondas de Ca2+.")
Participación de la fuga diastólica de Ca2+ en la generación de actividad automática. Imágenes representativas de los aumentos transitorios de Ca2+ producidos por estimulación eléctrica (0.5Hz, líneas verticales verdes) en cardiomiocitos control (A, control) y con fuga de Ca2+ anormal durante la diástole (B, actividad automática). Las células cargadas con el indicador Fluo-3 se escanearon en la modalidad de line-scan en el microscopio confocal (línea negra), lo que permite generar las imágenes bidimensionales donde el ejeX representa el tiempo en segundos y el ejeY representa la longitud de la célula en micrómetros. La amplitud del transitorio de Ca2+ intracelular (F/F0) producido por el estímulo eléctrico se muestra debajo de cada imagen. Las flechas rojas en las imágenes bidimensionales indican la fuga de Ca2+ durante la diástole en forma de chispas de Ca2+ y ondas de Ca2+.
Se ha propuesto que el aumento en la [Ca2+]i diastólica y/o en la [Ca2+]RS disminuyen el umbral de activación del RyR, favoreciendo la propagación de ondas de Ca2+ espontáneas mediante el proceso de activación-difusión-activación34,52,55. Incluso se ha considerado que algunos parámetros de las ondas de Ca2+, tales como la frecuencia, la amplitud y la velocidad de propagación, son predictivos de su potencial arritmogénico34,55,56. Sin embargo, paradójicamente existen enfermedades donde la actividad diastólica del RyR está aumentada aun en presencia de una [Ca2+]RS disminuida y que se explican por una mayor sensibilidad del RyR al Ca2+ luminal y/o citoplasmático, que favorecen su activación exacerbada9,57. En un estado de alteración crónica el incremento anormal de la fuga de Ca2+ no puede durar por siempre, ya que las células contienen poderosos mecanismos de autorregulación para controlar la actividad anormal de los RyR a largo plazo58. Desafortunadamente, los mecanismos moleculares específicos que participan en modificar la fuga de Ca2+ diastólica en ciertas enfermedades cardiacas aún no han sido completamente aclarados.
Función fisiológica de la fuga diastólica de Ca2+ mediada por el receptor de rianodinaLa participación del RyR en inducir una fuga de Ca2+ del RS se evidenció en un trabajo del grupo de Lakatta, en el que se observó que la incubación de cardiomiocitos intactos y en reposo con rianodina a concentraciones nanomolares vaciaba los reservorios intracelulares de Ca2+ localizados en el RS, lo cual se explica por la propiedad de este alcaloide de unirse únicamente a la conformación abierta del canal y de mantenerlo subconductante59. Posteriormente, Urthaler et al.60 reforzaron la idea de que el RyR fugaba Ca2+ en la diástole al mostrar que la RyR aumentaba esta fuga por inhibir el cerrado de los canales de Ca2+ del RS. Más tarde, Yamazawa et al.61 mostraron que la expresión de RyR2 en cultivos primarios de músculo esquelético de ratones dispédicos (es decir, que no expresan al RyR tipo1 o esquelético) inducía chispas de Ca2+ evocadas y ondas de Ca2+ espontáneas a [Ca2+]i basales, sugiriendo que la isoforma cardiaca del RyR se activa a [Ca2+]i diastólica y puede mediar la fuga de Ca2+. En 1998, Takeshima et al.62 proponen que durante el desarrollo embrionario los RyR2 se activan en la diástole en respuesta a elevaciones en la [Ca2+]RS, comportándose como válvulas de seguridad para prevenir la sobrecarga de Ca2+ del RS. Esta hipótesis la plantean al observar que en cardiomiocitos de embriones que carecen del RyR2 el Ca2+ se acumulaba gradualmente dentro de los reservorios del RS induciendo la formación de vacuolas a partir del RS, anormalidades en las mitocondrias y desregulación de la homeostasis del Ca2+, lo que conduce a una disfunción general del cardiomiocito62. Sin embargo, la función fisiológica de la fuga de Ca2+ mediada por el RyR no está restringida al corazón en desarrollo, también está presente en el corazón adulto y permite mantener la [Ca2+]RS dentro de un rango normal, como un mecanismo de balance a la actividad de recaptura de la bomba SERCA, permitiendo la estabilización de la actividad del RyR entre latido y latido63,64. En cardiomiocitos normales el incremento en la [Ca2+]RS aumenta la frecuencia de las chispas de Ca2+ espontáneas, permitiendo que el RyR2 regule el contenido de Ca2+ del RS a través de la fuga mediada por chispas65.
Arritmias relacionadas con alteraciones en la fuga diastólica de Ca2+Las arritmias cardiacas asociadas a enfermedades hereditarias o adquiridas son causantes de la morbimortalidad de un gran porcentaje de la población de los países industrializados, como Estados Unidos y México. De las 590,693 defunciones registradas en México en 2011, el 23.8% (140,595) corresponden a muertes relacionadas con enfermedades cardiovasculares (fuente INEGI), y entre ellas deben estar algunas arritmias, como la fibrilación ventricular.
En ciertas enfermedades cardiacas la fuga de Ca2+ está alterada y participa en la generación de arritmias adquiridas (como las que se presentan en la insuficiencia cardiaca, la miocardiopatía diabética y la fibrilación auricular) o hereditarias (como la taquicardia ventricular polimórfica y catecolaminérgica [TVPC] y la displasia arritmogénica del ventrículo derecho tipo 2 [DAVD2])66. En 2001, 2grupos de investigadores identificaron un total de 11mutaciones puntuales en el gen que codifica para el RyR cardiaco, el RYR2, asociadas con la presencia de TVPC en 7familias diferentes67,68 y a DAVD2 en 4familias independientes69. Hasta la fecha se han identificado 128variantes del hRYR2 y 12variantes de la calsecuestrina2 que están asociadas a estas arritmias (http://www.fsm.it/cardmoc/)70, lo que ha abierto un nuevo panorama al evidenciar que las alteraciones en la fuga de Ca2+ vía el RyR2 pueden ser parte de las bases moleculares de la arritmogénesis cardiaca.
Mutaciones del receptor de rianodina asociadas a taquicardia ventricular polimórfica y catecolaminérgicaLa TVPC es una arritmia hereditaria que se caracteriza por la aparición de una taquicardia ventricular que puede ser bidireccional o polimórfica inducida por estrés o ejercicio sin presentar alteraciones estructurales del corazón67,68,71. En la mayoría de los casos la taquicardia ventricular conduce a fibrilación ventricular y síncope o muerte súbita. Debido a que el electrocardiograma de los pacientes con TVPC no presenta anormalidades en condiciones de reposo, el diagnóstico implica la detección de la arritmia inducida por estrés durante una prueba de ejercicio o Holter. Esta arritmia hereditaria se presenta en 2formas: dominante y recesiva. La forma dominante de esta arritmia está asociada a mutaciones en el gen RYR2 que codifica al RyR cardiaco o tipo2 (RyR2), mientras que la forma recesiva está asociada alteraciones en el gen de la calsecuestrina2 (CSQ2), proteína que se encuentra en la luz del RS, que tiene una gran capacidad para unir Ca2+ y que participa junto con triadina y junctina en modular la respuesta del RyR al Ca2+ luminal72.
Debido a la importancia que tiene el determinar las bases moleculares de las arritmias, se han generado ratones transgénicos que expresan al RyR2 con alguna de las mutaciones puntuales encontradas en TVPC. El ratón knock in heterocigoto para la variante RyR2R4497C recapitula muchas de las características fenotípicas de los pacientes con TVPC, bajo estimulación β-adrenérgica73. Los cardiomiocitos ventriculares del ratón heterocigoto presentan DAD y actividad automática en presencia de isoproterenol9. En condiciones de reposo existe un aumento en la fuga de Ca2+ diastólica en forma de mayor frecuencia de chispas, el cual se ve exacerbado en presencia de isoproterenol, aun en ausencia de una sobrecarga de Ca2+ del RS9. La actividad anormal del RyRR4497C se ha explicado por un incremento exclusivo en su sensibilidad al Ca2+ luminal; sin embargo, esta idea es controversial, debido a que también existe evidencia de que su sensibilidad al Ca2+ del citoplasma esta aumentada, lo que le permite activarse más aun en condiciones de baja [Ca2+]RS9. Es posible, entonces, que las mutaciones del RyR2 asociadas con TVPC den lugar a varios fenotipos moleculares arritmogénicos, en lugar de un solo fenotipo (p.ej., incremento a sensibilidad a Ca2+ luminal). En apoyo a esta hipótesis, una mutación altamente arritmogénica del RyR2 asociada con TVPC, la RyR2V2475F, presenta una mayor sensibilidad al Ca2+ citoplasmático y luminal y también respuesta exacerbada a la fosforilación por la cinasa de proteínas dependiente de AMPc (PKA)74.
Fuga de Ca2+ en la insuficiencia cardiacaLa insuficiencia cardiaca (IC) continúa siendo un problema serio de salud en los países industrializados, como México. Las causas de la IC son muy heterogéneas, pero destacan las asociadas a la presencia de cardiomiopatías congénitas, hipertensión arterial, nefropatías, obesidad y diabetes mellitus, entre otras. Las consecuencias funcionales de la IC en la actividad del corazón se manifiestan como disfunción contráctil, actividad arritmogénica y remodelamiento estructural patológico. Parte de la disfunción del corazón en la IC se podría explicar por un aumento de la estimulación simpática y la concomitante liberación de catecolaminas que induce la activación de los receptores β-adrenérgicos; sin embargo, en los pacientes con IC crónica la respuesta β-adrenérgica se encuentra abatida. Una observación consistente es que los RyR2 aislados de corazones insuficientes, tanto de humanos como de modelos experimentales, presentan alteraciones en su actividad a [Ca2+]i en reposo, lo cual se refleja en un aumento de la fuga de Ca2+ diastólica54,75, aunque estas alteraciones no necesariamente se reflejan en cambios consistentes en la actividad del RyR en forma de chispas. Por el contrario, cardiomiocitos aislados de diversos modelos experimentales de IC muestran aumento75,76, disminución77 o ningún cambio evidente7 en la fuga de Ca2+ en forma de chispas. Las posibles explicaciones a estas discrepancias pueden encontrarse en diferencias específicas de cada modelo experimental de IC principalmente relacionadas con la modulación in situ del RyR. En estas diferencias encontramos modificaciones postraduccionales (como fosforilación, oxidorreducción y S-nitrosilación), aunado al remodelamiento estructural que altera la distribución de los RyR dentro de las unidades de liberación y que podría favorecer la fuga de Ca2+ vía ondas u oscilaciones de Ca2+ (fig. 2), o vía los RyR «rebeldes»46,54. En el año 2000 el grupo de A. Marks16 propuso que el aumento de los niveles de catecolaminas en los pacientes con IC activa a la PKA vía los receptores β-adrenérgicos, lo que induce la «hiperfosforilación» del RyR2 en la Ser2808 y la concomitante disociación de la FKBP12.6. En este esquema, estas modificaciones provocan la actividad descontrolada del RyR, lo que incrementa la fuga de Ca2+ diastólica y la propensión a eventos arrítmicos16,66. Sin embargo, diversos grupos de investigación han aportado evidencias que no permiten apoyar esta hipótesis32. Por ejemplo, el AMPc incrementa la frecuencia de chispas de Ca2+ en cardiomiocitos permeabilizados con estreptolisina-O, pero esto se debe a un aumento en la [Ca2+]RS (por el incremento en la actividad de la bomba SERCA) y no a un efecto directo sobre el RyR y su estado de fosforilación78. De hecho, la fosforilación del RyR en la Ser2808 no disocia a la FKBP12.6 ni modifica sustancialmente su actividad32; por el contrario, este sitio se ha encontrado constitutivamente fosforilado, independientemente de la presencia o no de IC. Además, estudios recientes en el modelo murino que expresa al RyRS2808A en tejido cardiaco —lo que evita la fosforilación de este residuo y por lo tanto, teóricamente, estabiliza la interacción RyR-FKBP12.6— muestran que los ratones RyRS2808A presentan una respuesta normal a la estimulación β-adrenérgica y una progresión similar a la IC comparado con los ratones que expresan al RyR2 normal79,80. Estos hallazgos controversiales requieren mucho trabajo adicional para aclarar la participación de la fosforilación del residuo Ser2808 en la fuga de Ca2+ diastólica durante la IC.
Adicionalmente, la activación de los receptores β-adrenérgicos también induce la fosforilación del RyR vía la cinasa de proteínas dependiente de Ca2+ y calmodulina tipo2 (CaMKII). Hasta el momento se han identificado 3sitios de fosforilación en el RyR2 con relevancia fisiológica: la Ser2808, que puede ser fosforilada por PKA o CaMKII; la Ser2814, al parecer exclusivamente fosforilada por CaMKII, y la Ser2030, únicamente fosforilada por PKA32, y no se tiene un consenso sobre cuál de estos sitios promueve el fenotipo que participa en la fuga de Ca2+ alterada. Trabajos muy recientes proponen que la fosforilación del residuo Ser2814 vía CaMKII es fundamental para inducir la fuga de Ca2+ alterada y la actividad arritmogénica en la IC81,82, por lo que se están acumulando más evidencias que muestran que es la actividad de la CaMKII y no la de la PKA la que favorece la actividad exacerbada del RyR durante la diástole y promueve la arritmogénesis en la IC.
Estrategias farmacológicas para el control de arritmias que involucran al receptor de rianodinaDebido a las evidencias que muestran que la fuga de Ca2+ diastólica vía los RyR está asociada a la generación de actividad arrítmica, diversos grupos de investigación han dedicado sus esfuerzos al desarrollo de estrategias terapéuticas para reducir la actividad anormal de los RyR83. Dentro de los fármacos utilizados encontramos inhibidores de los receptores β-adrenérgicos como el metoprolol y el carvediol84,85, derivados de hidantoínas como el dantroleno86-88, derivados del diltiazem como el JTV519 (o K201) y el S10789-92, agentes antiarrítmicos de la clase Ic, como la flecainida93-95, y glucósidos de resveratrol como la polidatina96; muchos de estos fármacos aún están en fase experimental debido a que existen diversas controversias sobre sus mecanismos de acción, como es el caso del JTV519, por lo que no han sido aprobados para el uso clínico.
Los bloqueadores de los receptores β-adrenérgicos disminuyen la fuga de Ca2+ vía el RyR2 al reducir la fosforilación del canal independientemente de si es mediada por PKA o CaMKII84, o bien por cambiar el estado redox del RyR2 al disminuir la oxidación de grupos tiol85.
El dantroleno es un relajante muscular utilizado como agente terapéutico en pacientes que padecen hipertermia maligna. El dantroleno se une en la región que corresponde a los aminoácidos 601 a 620 del RyR2 y que conecta los dominios N-terminal y central del canal, y se ha demostrado que corrige la actividad anómala del RyR2 en un modelo de IC en perro86. Adicionalmente el dantroleno disminuye la frecuencia de las chispas de Ca2+ espontáneas en cardiomiocitos del ratón que expresan el RyRR2474S87, así como en cardiomiocitos de conejo con IC, aunque en este último caso el dantroleno también indujo un aumento en la [Ca2+]RS, lo que podría favorecer una sobrecarga de Ca2+ del RS88.
El JTV519, también llamado K201, es un derivado de la 1-4 benzotiazepina que ha sido propuesto como un posible agente terapéutico para disminuir la fuga de Ca2+ diastólica vía el RyR2 en la IC y en la TVPC al reducir la disociación de la proteína FKPB12.689,90,97. Sin embargo, existen trabajos que muestran que el JTV519 no disminuye la aparición de DAD en un modelo murino de TVPC98, además de que su mecanismo de acción es independiente del nivel de fosforilación del RyR o de la cantidad de FKBP12.6 que tenga asociada91,92. Adicionalmente, el JTV519 inhibe otros canales iónicos que se expresan en el tejido cardiaco99,100, por lo que al tener efectos pleotrópicos su uso como agente antiarrítmico es cuestionable.
La flecainida es un agente antiarrítmico del tipo Ic debido a que su mecanismo de acción involucra la inactivación de los canales de Na+ dependientes de voltaje (INa). Recientemente se encontró que la flecainida también reduce la fuga de Ca2+ diastólica en un modelo murino de TVPC94, aunque existen opiniones divergentes acerca de si el fármaco actúa directamente en el RyR o bien modula su actividad de forma indirecta al inhibir la INa95. Incluso existen evidencias de que la flecainida reduce la masa de la chispa de Ca2+ pero a la vez incrementa su frecuencia, por lo que el efecto neto en la fuga de Ca2+ mediada por chispas sería nulo93.
Recientemente se introdujo la polidatina como un posible agente cardioprotector. La polidatina es un glucósido del resveratrol aislado de la planta Polygonum cuspidatum con potentes efectos antioxidantes. Se ha demostrado que disminuye la fuga de Ca2+ mediada por chispas al reducir los niveles de especies reactivas de oxígeno y restaurar los grupos tioles del RyR2 en cardiomiocitos de animales lesionados por quemaduras96. Sin embargo su introducción es muy reciente, por lo que se requieren más estudios para evaluar posibles efectos inespecíficos.
Es importante aclarar que no toda la fuga de Ca2+ diastólica ocurre vía los RyR, ya que existen evidencias de que puede haber otros canales iónicos involucrados. Como ejemplo, recientemente se encontró que la proteína PLB en su forma fosforilada puede aumentar la fuga pasiva de Ca2+ del RS, la cual es resistente al efecto del rojo de rutenio101.
Por último, las estrategias terapéuticas futuras deberán evaluar que los posibles fármacos antiarrítmicos no interfieran con la liberación sistólica de Ca2+ en condiciones fisiológicas y que, a su vez, la fuga diastólica no sea completamente suprimida debido a su participación tan importante en la regulación de la [Ca2+]RS.
FinanciaciónEste trabajo fue apoyado por el Instituto de Ciencia y Tecnología del Distrito Federal (ICyTDF, proyecto No.331/2010) para A. R. y el Instituto Nacional de Salud de EE.UU. (NIH, donativos HL055438 y HL094291) para H.H.V.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.