


A hiperplasia congénita da suprarrenal não clássica (HCSRNC) é uma das doenças autossómicas recessivas mais frequentes que, na grande maioria dos casos, ocorre por deficiência da enzima 21‐hidroxilase (21‐OH). Com o presente artigo pretende‐se fazer uma revisão sobre os aspetos relevantes desta doença na prática clínica diária, em especial nas áreas da pediatria, endocrinologia e obstetrícia.
MétodosFoi efetuada uma revisão bibliográfica através da base de dados eletrónica MEDLINE de artigos publicados entre 1990‐2013. Foram utilizadas as palavras‐chave: «hiperplasia da suprarrenal não clássica» e «deficiência de 21‐hidroxilase».
ConclusõesA apresentação clínica da HCSRNC é variável, desde a ausência completa de sintomas até à existência de um ou vários sintomas relacionados com o excesso de androgénios, como pubarca precoce, hirsutismo, oligomenorreia, acne ou infertilidade. O diagnóstico é confirmado através da prova de estimulação com tetracosactídeo. O tratamento deve ser efetuado apenas se houver sintomatologia, deve ser individualizado e tem como objetivos principais evitar uma puberdade precoce com encerramento epifisário prematuro, regularizar os ciclos menstruais, promover a fertilidade e atenuar o acne e hirsutismo.
Nonclassical congenital adrenal hyperplasia is one of the most common autosomal recessive disorders and, in the majority of cases, is due to 21‐hydroxylase (21‐OH) enzyme deficiency. With this article we intend to review the relevant aspects of this disease in daily clinical practice, particularly in the areas of pediatrics, endocrinology and obstetrics.
MethodsA bibliographic review was performed through the electronic database MEDLINE, for articles published between 1990 and 2013. The keywords used were “nonclassical adrenal hyperplasia” and “21‐hydroxylase deficiency.”
ConclusionsThe clinical presentation of nonclassical adrenal hyperplasia is variable: it can be asymptomatic or cause symptoms related to androgen excess as premature pubarche, hirsutism, oligomenorrhea, acne or infertility. The diagnosis is confirmed by a tetracosactide stimulation test. Treatment is reserved for symptomatic individuals, must be individualized and its main goals are to avoid premature pubarch with premature epiphyseal fusion, to regulate the menstrual cycles, to promote fertility and to reduce hirsutism and acne.
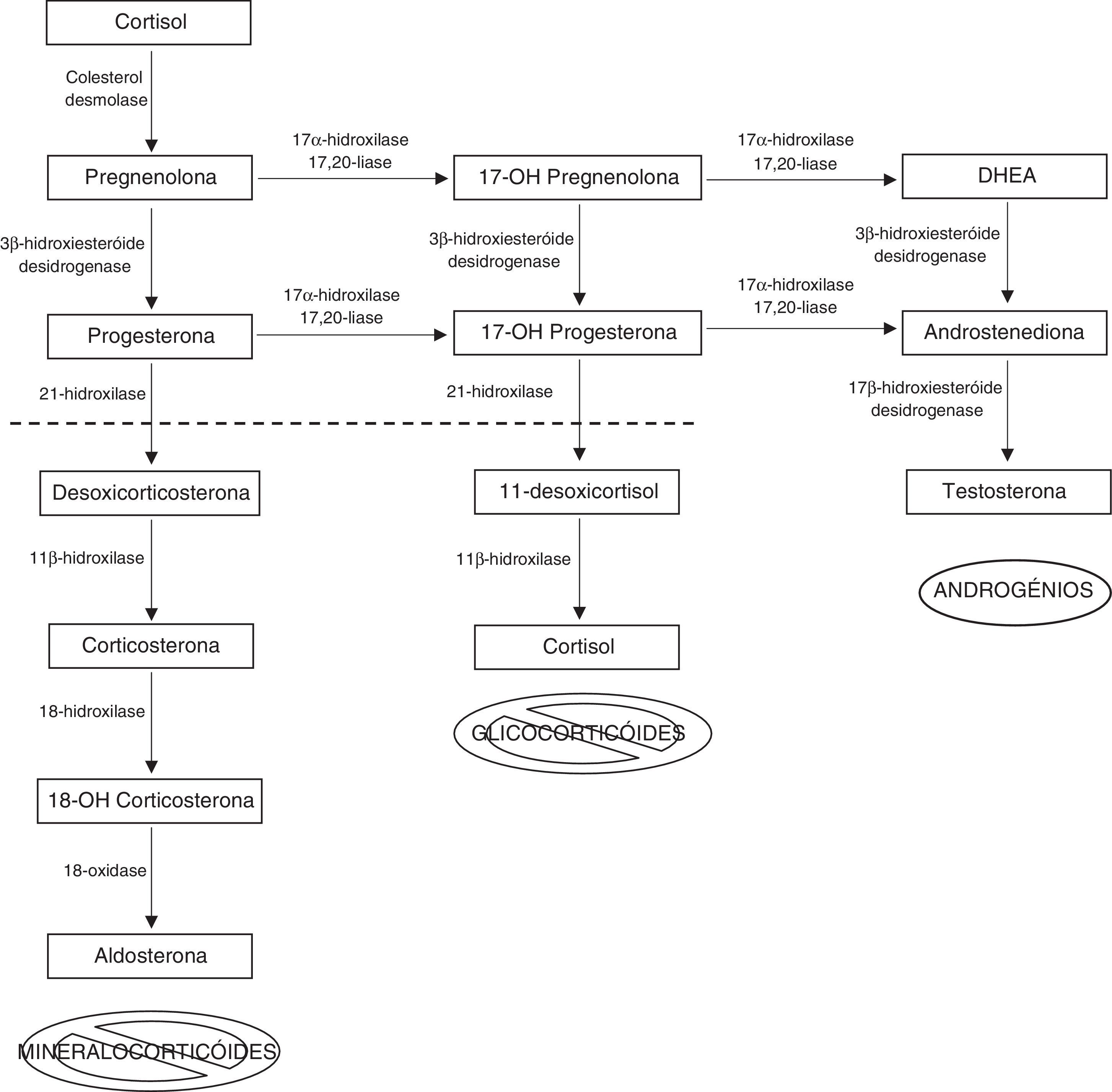
A hiperplasia congénita da suprarrenal (HCSR) consiste num grupo de doenças genéticas autossómicas recessivas em que uma ou mais etapas da biossíntese do cortisol estão afetadas, devido a mutações nos genes que codificam as enzimas envolvidas na esteroidogénese1. Uma síntese insuficiente de cortisol conduz a um aumento de hormona libertadora de corticotropina (CRH) e de hormona adrenocorticotrópica (ACTH), o que provoca estimulação do córtex da suprarrenal, levando à sua hiperplasia2. Cerca de 90‐95% dos casos são causados por deficiência da enzima 21‐hidroxilase (21‐OH)3. Esta enzima é responsável pela conversão de 17‐hidroxiprogesterona (17‐OHP) em 11‐desoxicortisol e é codificada pelo gene CYP21A21. A gravidade da doença depende do tipo de mutação da enzima 21‐OH4. Verifica‐se acumulação dos precursores hormonais a montante do defeito enzimático como é o caso da 17‐OHP. Estes precursores são desviados para a via da síntese de androgénios, que não necessita da 21‐OH, levando a níveis elevados de androstenediona, testosterona, dihidrotestosterona e estrogénios aromatizados perifericamente2,3 (fig. 1). Nas formas mais graves de HCSR, «formas clássicas», há défice de cortisol e de aldosterona em 100 e 75% dos casos, respetivamente, e o seu diagnóstico é efetuado logo no período neonatal ou na pequena infância com insuficiência suprarrenal. A prevalência global de HCSR clássica é de 1:10.000 a 1:18.000 nascimentos (varia de 1:280 nos esquimós Yupic até 1:28.000 na China)1,5. A incidência é semelhante em homens e mulheres, contudo, nos doentes do sexo masculino o diagnóstico raramente é efetuado à nascença4. Nas formas mais leves, «formas não clássicas», a atividade enzimática está diminuída, contudo, é suficiente para manter uma produção glicocorticoide e mineralocorticoide adequadas, ainda que se verifique um aumento de produção de androgénios, o que lhes confere um início mais tardio com sinais de hiperandrogenismo ou podem mesmo ser assintomáticas. A sua prevalência global é de cerca de 1:1.0003.
Epidemiologia
A HCSRNC é uma das doenças autossómicas recessivas mais frequentes, estando sem dúvida subdiagnosticada, especialmente nos homens4. A sua prevalência varia de acordo com a raça e etnia, sendo de cerca de 0,1% na população geral, 1% em Nova Iorque, 1‐2% nos hispânicos e 3‐4% nos judeus Ashkenazi (Europa de Leste)1,3,4,6.
Nas mulheres com hiperandrogenismo estima‐se que a prevalência de HCSRNC seja de cerca de 2%, variando entre 0,6‐9,0%, dependendo das séries7–10. A percentagem de crianças com puberdade precoce que tem HCSRNC é incerta, com descrição desde prevalências muito baixas até valores de 30% em grupos de alto risco3. Também é desconhecida a prevalência de HCSRNC em homens com oligospermia.
Existe uma elevada frequência de portadores assintomáticos: 10% na forma não clássica e 1,6% na forma clássica3,11.
Devido ao estigma e ansiedade provocados pelo diagnóstico de uma doença genética, alguns autores sugerem que a forma não clássica de HCSR só deve ser catalogada como doença genética no caso de apresentar sinais de excesso de androgénios, caso contrário deve ser encarada como um polimorfismo genético3.
GenéticaOs seres humanos têm 2 genes CYP21A: um pseudogene não funcional (CYP21P ou CYP21A1) e um gene ativo (CYP21 ou CYP21A2), ambos localizados no braço curto do cromossoma 612. A mutação em causa é que vai condicionar a percentagem de atividade enzimática da 21‐OH que, por sua vez, determina a gravidade da doença. Nas formas não clássicas, a 21‐OH apresenta cerca de 20‐50% da sua atividade3,4. Assim, o fenótipo é variável dependendo do grau de défice enzimático, existindo uma boa correlação entre o genótipo e o fenótipo, documentada em cerca de 98% dos casos3,4,12. Existem muitos polimorfismos no gene CYP21 com normal função enzimática. A mutação missense V281L é a mais frequentemente associada à HCSRNC. Outras mutações associadas a esta patologia são as mutações missense P30L, P453S e R339H1,4,10.
A maioria dos doentes são heterozigotos compostos, ou seja, apresentam mutações diferentes em cada um dos alelos, sendo a forma clínica determinada pelo alelo com maior atividade enzimática12. Os portadores apresentam um alelo normal e um alelo mutado e não têm doença clínica, ainda que possam apresentar alterações bioquímicas discretas3.
O facto de a correlação genótipo‐fenótipo não ser a 100% sugere a influência de outros genes e do ambiente no aparecimento de manifestações clínicas. A variação interindividual na biossíntese e sensibilidade aos androgénios também tem importância nas manifestações clínicas da doença3.
Manifestações clínicasA HCSRNC tem habitualmente um início tardio com aparecimento de sinais de hiperandrogenismo no final da infância, na adolescência ou no início da idade adulta. A sua apresentação clínica é muito variável, desde a ausência completa de sintomas até à evidência de um ou vários sinais de excesso de androgénios4. Os sinais que devem alertar o clínico para esta patologia são: pubarca precoce, idade óssea avançada, hirsutismo, acne, oligomenorreia ou infertilidade1. Não existem alterações hidroeletrolíticas uma vez que não há défice das linhas glicocorticoide e mineralocorticoide13.
Nas crianças, podem ocorrer sinais de hiperandrogenismo como pubarca prematura, acne e crescimento acelerado com elevada estatura na infância, com idade óssea avançada (+2,0 desvios‐padrão para idade e sexo) com encerramento precoce das epífises, o que vai condicionar baixa estatura na idade adulta3,4,14. Não se verifica ambiguidade sexual.
Nas mulheres, as manifestações clínicas variam de acordo com a idade. Na infância podem ocorrer as manifestações clínicas anteriormente referidas para as crianças. Na adolescência e idade adulta, de acordo com alguns estudos, pode ocorrer hirsutismo (59%), oligomenorreia (54%), acne (33%), infertilidade (13%), clitoromegalia (10%), alopecia (8%) ou amenorreia primária (4%)10,15–17. O risco de abortamento espontâneo parece ser maior nas mulheres com HCSRNC (26%) do que nas mulheres sem esta patologia15. Cerca de 50% das mulheres com HCSRNC necessitam de terapêutica glicocorticoide para engravidar2,15. É muitas vezes difícil fazer o diagnóstico diferencial com a síndrome do ovário poliquístico, uma vez que a apresentação clínica pode ser similar8.
Nos doentes do sexo masculino os órgãos genitais são normais; pode ocorrer puberdade precoce e acne e verifica‐se um crescimento somático muito rápido com maturação precoce do esqueleto, o que pode condicionar baixa estatura final1. A maioria dos doentes tem função testicular e fertilidade normais, podendo uma pequena percentagem apresentar testicular adrenal rest tumors (TART) ou oligospermia com infertilidade associada3.
Não há consenso quanto ao facto de, se um indivíduo ser portador heterozigoto, tal implicar um maior risco de desenvolver sintomas de hiperandrogenismo. Um estudo dinamarquês documentou que a prevalência de portadores heterozigotos era de 8,6% em 252 mulheres com hirsutismo e de 6,3% em 252 mulheres sem hirsutismo18.
DiagnósticoQuando há suspeita clínica de HCSRNC deve ser efetuado o doseamento de 17‐OHP basal pela manhã (por volta das 8 horas da manhã). Nas mulheres é conveniente efetuar este doseamento no início da fase folicular do ciclo menstrual, uma vez que o corpo lúteo produz 17‐OHP podendo originar falsos positivos se a colheita for feita na fase lútea do ciclo7. Este rastreio poderá também ter interesse em indivíduos com história familiar de HCSR e em mulheres com elevado risco de doença atendendo à sua etnia4. Alguns autores defendem a realização do rastreio de HCSRNC no período pré‐conceção em mulheres com sinais de hiperandrogenismo e dificuldade em engravidar, uma vez que é muito mais económico diagnosticar e tratar esta patologia do que efetuar um conjunto de estudos complementares associados à infertilidade e eventualmente realizar algumas técnicas e tratamentos de infertilidade que são muito dispendiosos4.
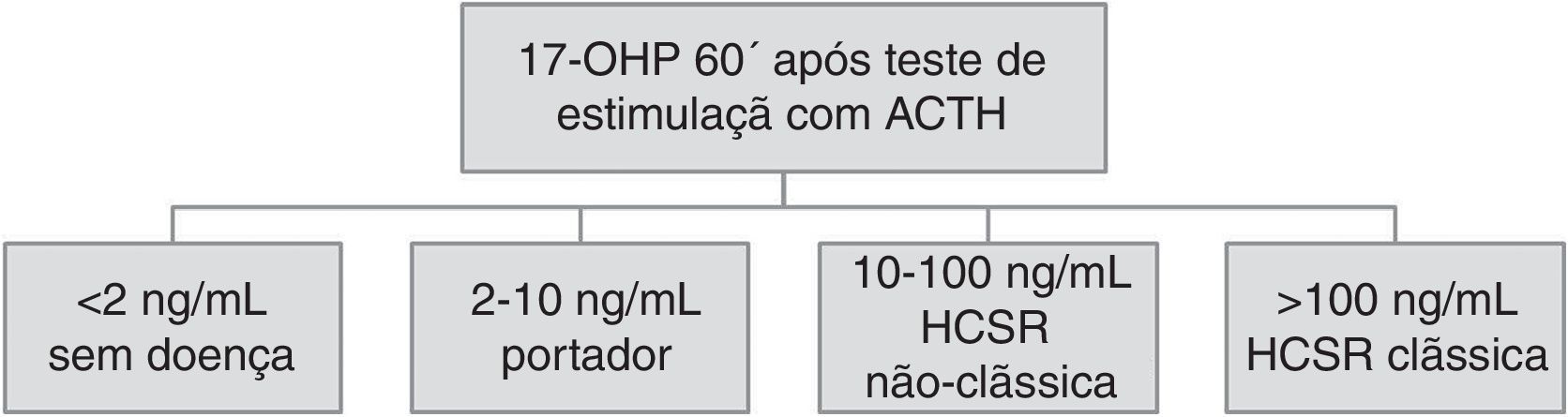
Nos indivíduos com elevação da 17‐OHP basal, o diagnóstico de HCSR deve ser confirmado através de um teste de estimulação com tetracosactídeo (ACTH sintética). Assim, valores de 17‐OHP pela manhã superiores a 2ng/mL (6nmol/L) ou superiores a 0,82ng/mL (2,5nmol/L) na infância são sugestivos de existência de doença1,3. Nestes casos deve ser efetuado o teste de estimulação com ACTH, que consiste no doseamento de 17‐OHP basal e 60 minutos após injeção endovenosa de 250μg de ACTH. Valores de 17‐OHP aos 60 minutos entre 2‐10ng/mL (6‐30nmol/L) podem corresponder a portadores heterozigotos, valores entre 10‐100ng/mL (30‐300nmol/L) são sugestivos de HCSRNC e valores superiores a 100ng/mL (300nmol/L) são sugestivos de forma clássica da doença1,3,19 (fig. 2). Podem ainda estar presentes outras alterações analíticas, nomeadamente elevação de progesterona, 17‐hidroxipregnenelona, androstenediona, testosterona, assim como excreção urinária aumentada de metabolitos da 17‐OHP como o pregnanetriol3.

Este ponto de corte de 2ng/mL (6nmol/L) de 17‐OHP basal, para haver indicação para fazer o teste de estimulação com ACTH, e o valor de corte de 10ng/dL (30nmol/L) de 17‐OHP após a injeção de ACTH para ser diagnóstico de HCSRNC, foram obtidos antes de haver estudos genéticos e têm sido muito contestados. Um estudo brasileiro sugere que o teste com ACTH deve ser realizado quando a 17‐OHP basal for superior a 3,5ng/mL (10,5nmol/L), uma vez que este apresenta alta sensibilidade e bom valor preditivo negativo7. Com a era dos estudos moleculares verificou‐se uma sobreposição entre os níveis de 17‐OHP após ACTH encontrada em indivíduos heterozigotos para mutações da forma clássica e doentes com o diagnóstico da forma não clássica12.
Nas crianças recomenda‐se fazer o perfil adrenocortical completo após o teste de estimulação com ACTH para diferenciar a deficiência de 21‐OH de outros defeitos enzimáticos1.
Sugere‐se fazer o estudo genético para efeitos de aconselhamento genético e também nos casos em que o teste de estimulação com ACTH é duvidoso, ou seja, quando se verificam respostas «borderline» com valores de 17‐OHP entre 10‐17ng/mL (30‐51nmol/L) após estimulação com ACTH, com o objetivo de estes indivíduos não serem falsamente rotulados de HCSRNC, quando podem apenas corresponder a portadores heterozigotos12. Como o estudo molecular não está disponível em todos os centros, nestes casos difíceis os parâmetros clínicos de excesso de androgénios como avanço de idade óssea, clitoromegalia, hirsutismo acentuado ou infertilidade devem ser tidos em conta para introdução ou não de terapêutica3.
Devido à variação na concentração hormonal condicionada pelo ritmo circadiano, ao fazer apenas o doseamento basal de 17‐OHP pode ser perdido o diagnóstico, razão pela qual o teste de estimulação com ACTH deve ser efetuado sempre que haja forte suspeita do diagnóstico, mesmo que o doente apresente valores basais baixos de 17‐OHP (inferiores a 2ng/mL)7,19.
Os indivíduos portadores heterozigotos podem apresentar o mesmo tipo de alterações bioquímicas, mas com valores de 17‐OHP menos elevados e pequenas respostas ao teste de estimulação com ACTH (valores 17‐OHP aos 60 minutos entre 2‐10ng/mL, ou seja, 6‐30nmol/L)1.
TratamentoAs crianças com HCSRNC podem beneficiar da terapêutica com glicocorticoides caso se verifique pubarca precoce com idade óssea avançada, com alto risco de fusão epifisária precoce que condicione baixa estatura final1,3. O tratamento consiste em hidrocortisona em baixas doses. Se as crianças são assintomáticas não há indicação para tratamento. A introdução de terapêutica com o intuito de promover o crescimento não é consensual e só poderá, eventualmente, ser efetuada quando a estatura é ‐2,25 desvios‐padrão para idade e sexo1. Alguns estudos relatam a utilização de hormona do crescimento (GH) e de análogos de LHRH em crianças com HCSRNC com idade óssea avançada, com melhoria na estatura final alcançada em relação à esperada antes de iniciar o tratamento20.
Nas mulheres, são indicações para tratamento a existência de hirsutismo, acne, oligomenorreia ou infertilidade1,3. No período pré‐conceção poderá também ser efetuada terapêutica com glicocorticoides uma vez que esta parece estar associada a uma diminuição do risco de abortamento espontâneo15,21. No caso de não se pretender uma gravidez, a terapêutica de 1.a linha consiste na utilização de estroprogestativo e/ou antiandrogéneo, atendendo aos efeitos laterais dos glicocorticoides e ao facto de o hirsutismo necessitar de um tratamento prolongado13,22. No caso de a fertilidade ser desejada está indicado efetuar terapêutica com glicocorticoide e, se necessário, adicionar citrato de clomifeno ou outras técnicas de reprodução nos casos de oligomenorreia ou anovulação3,15,23. A terapêutica com glicocorticoides deve ser na menor dose possível para obter resultados13, sendo habitualmente 10‐20mg/dia de hidrocortisona em 2 administrações por dia, 0,25‐0,75mg/dia de dexametasona à hora de deitar ou 5‐7,5mg/dia de prednisolona à hora de deitar4,15. Não existe consenso no que diz respeito ao tipo de glicocorticoide a usar nem quanto à sua dose. Cerca de 36% dos endocrinologistas europeus utilizam hidrocortisona, 33% dexametasona (este é o fármaco habitualmente usado na pré‐conceção para induzir ovulação) e 14% prednisolona1. Se a gravidez for obtida sob corticoterapia, a sua manutenção durante a gestação deve ser ponderada caso a caso: poderá ser reduzida gradualmente até à sua suspensão (o que vai diminuir a sua iatrogenia) ou poderá eventualmente ser mantida quando a doente já fazia glucocorticoides previamente à gravidez e não apenas na indução da ovulação. Não deve ser utilizada a dexametasona durante a gravidez uma vez que esta atravessa a placenta, exceto quando se pretende suprimir a função suprarrenal do feto (nos casos de risco de descendência do sexo feminino com forma clássica da doença)13. O tratamento com glicocorticoide pode ser interrompido quando a mulher já não pretende novamente engravidar, contudo, se os sintomas de hiperandrogenismo persistirem, a doente pode beneficiar de efetuar um estroprogestativo3. O tempo para regressão dos sintomas pode ser de 3 meses no caso de acne ou irregularidades menstruais, mas pode demorar mais de 2 anos nos casos de hirsutismo1,4. Será também importante associar medidas cosméticas para obter bons resultados no tratamento do hirsutismo1,23. A terapêutica antiandrogénica pode também ser utilizada em casos de alopecia androgénica em doentes do sexo feminino10.
Nos doentes do sexo masculino o tratamento da HCSRNC é desnecessário, exceto se existir oligospermia e desejo de fertilidade ou se houver evidência de TART’, o que é muito raro na forma não clássica1,6. A posologia do tratamento glicocorticoide é similar à descrita para os doentes do sexo feminino, contudo, também não há consenso de qual será o fármaco ideal ou a dose mais adequada, sendo necessários mais estudos. O tratamento pode ser interrompido quando o doente não quiser ter mais filhos.
Monitorização do tratamento nos adultosOs adultos com HCSRNC devem ser avaliados clínica e analiticamente pelo menos uma vez por ano1.
Nos doentes do sexo feminino o objetivo é normalizar os níveis de androgénios no sentido de obter uma regularização dos ciclos menstruais e uma diminuição dos sinais e sintomas de hiperandrogenismo. Os objetivos laboratoriais consistem em normalizar os níveis de androstenediona e testosterona1,10; a normalização dos níveis de 17‐OHP normalmente significa um excesso de glicocorticoides, exceto nos casos de indução da ovulação10.
Nos doentes do sexo masculino a monitorização bioquímica do tratamento deve ser efetuada com doseamentos de 17‐OHP que deve estar ligeiramente acima do normal ou suprimida se existência de TART’, de androstenediona que deve estar ligeiramente elevada, de testosterona e de gonadotrofinas que devem ser normais1. Os níveis de testosterona refletem predominantemente a função gonadal e não a função suprarrenal, sendo preferível dosear a androstenediona na monitorização do tratamento1.
Não são necessárias doses de glicocorticoides de stress, exceto se houver supressão iatrogénica do eixo hipotálamo‐hipófise‐adrenal. É importante vigiar a densidade mineral óssea1,10.
Aconselhamento genéticoSerá importante efetuar aconselhamento genético quando o casal já teve um filho com HCSR ou quando um dos progenitores apresenta a doença na sua forma clássica ou não clássica1. Deve ser explicado que a HCSR é uma doença autossómica recessiva. No caso de o casal já ter tido um filho com HCSR, a probabilidade de ter outro feto com HCSR é de 25% (1/4) e a probabilidade de ter um feto do sexo feminino com HCSR é 12,5% (1/8). No caso de um dos progenitores ter HCSR forma clássica e, considerando uma frequência de portadores de HCSR na população geral de 1,6%, a probabilidade de ter um feto do sexo feminino com HCSR é de 0,4% ([100% de probabilidade de um progenitor ser portador de 2 alelos com mutação clássica] x [1,6% de probabilidade do outro progenitor ser portador de alelo com mutação clássica que é igual à da população em geral] x [50% de probabilidade de progenitor portador transmitir o alelo mutado] x [50% de probabilidade de o feto ser do sexo feminino]). No caso de um dos progenitores ter HCSR forma não clássica, atendendo ao facto de que pelo menos cerca de metade destes doentes apresentam o outro alelo com mutação que causa HCSR clássica10,15, a probabilidade de ter um feto do sexo feminino com HCSR é de 0,1% ([50% de probabilidade de o doente ter o outro alelo com mutação clássica] x [1,6% de probabilidade do outro progenitor ser portador de alelo com mutação clássica que é igual à da população em geral] x [25% de probabilidade de ambos os alelos com mutação clássica serem transmitidos para o feto] x [50% de probabilidade de o feto ser do sexo feminino])3. Nestas 2 últimas situações, seria benéfico efetuar o estudo genético ao outro progenitor21.
Diagnóstico pré‐natalO diagnóstico pré‐natal de HCSR pode ser efetuado através do estudo genético a partir de tecido das vilosidades coriónicas (obtido por biópsia entre as 9‐11 semanas de gestação) ou de células do líquido amniótico (obtidas por amniocentese entre as 15‐18 semanas de gestação), contudo, o estudo do gene CYP21 não deteta todas as mutações e não está sempre disponível3,17. O diagnóstico pré‐natal pode ainda ser realizado através do doseamento de 17‐OHP no líquido amniótico colhido por amniocentese, contudo, este método não pode ser usado em mães que estejam medicadas com dexametasona por esta suprimir o córtex adrenal do feto, a não ser que este tratamento seja interrompido cerca de 5‐7 dias antes da amniocentese3.
Terapêutica pré‐natalUma vez que a terapêutica pré‐natal apresenta risco de malformações do feto e iatrogenia não desprezível na grávida, recomenda‐se que esta seja utilizada apenas em situações muito específicas e de acordo com os protocolos de cada centro1,24. Esta terapêutica provoca bloqueio da produção adrenal do feto, estando reservada apenas para gestações com risco de feto com hiperplasia adrenal congénita clássica do sexo feminino com o objetivo de evitar virilização ou ambiguidade genital. Contudo, é importante informar os progenitores de que esta terapêutica não evita a necessidade de efetuar tratamento pós‐natal nos bebés com hiperplasia congénita da suprarrenal, ou seja, não evita o aparecimento da doença. O tratamento consiste na administração à grávida de dexametasona, uma vez que esta atravessa a placenta, na dose de 20ug/kg/dia (de acordo com o peso pré‐gestacional), num máximo de 1,5mg/dia dividida em 3 administrações, devendo ser iniciada antes da 8.a semana de gestação3. O sucesso é alcançado em 80‐85% dos casos1 e os motivos de insucesso são essencialmente um início tardio do tratamento, a falta de adesão ou a dose subterapêutica. A dexametasona deve ser interrompida quando o feto é do sexo masculino ou quando o diagnóstico pré‐natal exclui a forma clássica da doença4. Esta terapêutica apresenta riscos para o feto nomeadamente malformações congénitas como hipertrofia dos septos cardíacos ou fendas a nível orofacial1. Cerca de 10% das mulheres grávidas submetidas a esta terapêutica podem apresentar síndrome de Cushing iatrogénica, ganho ponderal excessivo, hipertensão arterial ou diabetes gestacional1,10.
Rastreio bioquímico neonatalO rastreio de HCSR é efetuado através do doseamento de 17‐OHP entre as 48‐72 horas de vida. A prematuridade, o baixo peso e as patologias neonatais estão associados a falsos positivos e as formas mais leves de HCSR associam‐se a falsos negativos como é o caso das formas não clássicas1,3. Está recomendada a utilização de um protocolo com 2 níveis, que consiste num imunoensaio em papel de filtro, seguido de um teste de confirmação no caso de resultados positivos para o primeiro teste25. Este rastreio apresenta cerca de 98% de sensibilidade e apenas 2% de especificidade, sendo os rastreios positivos abordados de acordo com a região. A Sociedade Europeia de Endocrinologia recomenda a realização de rastreio neonatal em todos os recém‐nascidos1, o que iria permitir diagnosticar a forma perdedora de sal que é potencialmente letal, especialmente nos rapazes, uma vez que não apresentam ambiguidade genital e permitir também diagnosticar as formas virilizantes e tratá‐las atempadamente. Quando se obtém um rastreio positivo é iniciada terapêutica e o bebé é enviado para observação por uma unidade de endocrinologia pediátrica, uma vez que tem indicação fazer teste de estimulação com ACTH e estudo de outras hormonas para além da 17‐OHP, nomeadamente cortisol, desoxicorticosterona, 11‐desoxicortisol, 17OH‐pregnenolona, DHEA e androstenediona1. A realização deste rastreio associado ao teste de estimulação com ACTH poderá também ser benéfica nos filhos de mães com HCSRNC uma vez que parece que a prevalência de situações de HCSR será bem superior à estimada matematicamente (2,5 vs 0,2%)15 e também porque irá permitir o diagnóstico precoce de situações de HCSR clássica e não clássica e assim estar alerta para os seus sinais e sintomas1.
ConclusãoA HCSRNC é uma doença autossómica recessiva comum e manifesta‐se habitualmente na infância tardia, adolescência ou início da vida adulta. Os sintomas típicos são pubarca prematura, hirsutismo, oligomenorreia, acne e infertilidade. Pode também ser assintomática. Apesar de ser uma doença genética, o seu diagnóstico é essencialmente laboratorial através do doseamento basal de 17‐OHP e da realização do teste de estimulação com ACTH. O tratamento só é necessário caso haja sintomatologia e deve ser individualizado de acordo com o sexo e idade do doente e também atendendo ao desejo ou não de uma gravidez. Este não se resume apenas à utilização de glicocorticoides, podendo também consistir no uso de uma pílula estroprogestativa, de medicação antiandrogénica ou de medidas cosméticas. O rastreio bioquímico neonatal habitualmente não deteta casos de HCSRNC.
Conflito de interessesOs autores declaram não haver conflito de interesses.
