




Molecular markers are increasingly being utilized in diagnosing, prognosticating and predicting response to therapy of gliomas. The 4th edition of the World Health Organization (WHO) Classification of Tumours of the Central Nervous System (CNS) was updated in 2016 and incorporates multiple molecular markers in combination with histology to arrive at an integrated pathological diagnosis1. Newer entities were defined and some were removed based on biological and clinical relevance. Clinical trials have retrospectively incorporated molecular markers and reported patient outcomes based on them, which have showed significant survival differences for specific subtypes of gliomas. Challenges with respect to interobserver variability in diagnosis based on histology alone or availability of minimal diagnostic brain tissue can now be addressed with the use of molecular markers.
The WHO 2016 classification integrated genotypic and phenotypic parameters that add to diagnostic accuracy of CNS tumors. This in turn will facilitate more accurate determination of prognosis and development of targeted treatments. An algorithm beginning with histology and followed by addition of molecular testing for diagnosing different subtypes of gliomas has been suggested. Isocitrate dehydrogenase (IDH) mutation testing either by immunohistochemistry or sequencing will now be part of routine testing and will classify gliomas as IDH-mutant or -wildtype. As shown in several studies, IDH-mutant gliomas are generally associated with good response to treatment and a better prognosis2,3. Oligoastrocytoma grade II or III as a diagnosis has been a subject of high interobserver variability4,5, which can now be better designated as either oligodendroglioma or astrocytoma with the use of molecular markers IDH, ATRX, and chromosomes 1p and 19q. However, given the chance of discordant results and the non-availability of molecular testing, these diagnostic entities are kept in the new classification system as oligoastrocytoma and anaplastic oligoastrocytoma NOS (not otherwise specified), although in general this diagnosis is being discouraged. The NOS designation has also been added as a possibility to grades II and III oligodendrogliomas and astrocytomas and grade IV glioblastomas. The use of NOS is to be used in two situations: if molecular testing has not been performed or if molecular testing was not conclusive. Gliomatosis cerebri, an entity that was defined in the original WHO 4th edition from 20076, has now been deleted from the 2016 classification as it indicates a pattern of spread better defined on imaging. It is reportedly seen in IDH-mutant astrocytoma and oligodendroglioma as well as IDH-wildtype glioblastoma7,8. Two diffuse astrocytoma variants, namely protoplasmic and fibrillary astrocytomas have also been deleted from the new classification; however, gemistocytic astrocytoma has kept its defined place, but only as an IDH-mutant subtype. This once again shows the importance of associated molecular component in making the diagnosis. Diffuse midline glioma characterized by a K27M mutation in the histone H3 genes H3F3A or HIST1H3B/C is a newly defined entity. These tumors have a diffuse growth pattern, have midline location, occur in both children and adults, and are associated with a very poor prognosis1. Diffuse intrinsic pontine glioma (DIPG) is considered a part of this new entity. This molecular definition will facilitate development of therapies directed against this mutation. Other molecular markers in non-glioma tumors that have now been incorporated in the new WHO classification include RELA fusion-positive supratentorial ependymoma, mostly occurring in children; C19MC-altered embryonal tumors with multilayered rosettes; and restructuring of medulloblastomas based on WNT, SHH (sonic hedgehog), and TP53 mutations. For the purpose of this review we will focus on molecular markers in diffuse gliomas only (Table 1).
CHANGES IN CLASSIFICATION OF GLIOMAS BETWEEN 2007 AND 2016 WHO CLASSIFICATION SYSTEMS
| WHO 2007 | WHO 2016 |
|---|---|
| Diffuse astrocytoma | Diffuse astrocytoma, IDH-mutant |
| Gemistocytic astrocytoma, IDH-mutant | |
| Diffuse astrocytoma, IDH-wildtype | |
| Diffuse astrocytoma, NOS | |
| Anaplastic astrocytoma | Anaplastic astrocytoma, IDH-mutant |
| Anaplastic astrocytoma, IDH-wildtype | |
| Anaplastic astrocytoma, NOS | |
| Glioblastoma | Glioblastoma, IDH-wildtype |
| Glioblastoma, IDH-mutant | |
| Glioblastoma, NOS | |
| Oligodendroglioma | Oligodendroglioma, IDH-mutant and 1p/19q-codeleted |
| Oligodendroglioma, NOS | |
| Anaplastic oligodendroglioma | Anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted |
| Anaplastic oligodendroglioma, NOS | |
| Oligoastrocytoma | Oligoastrocytoma, NOS |
| Anaplastic oligoastrocytoma | Anaplastic oligoastrocytoma, NOS |
| Did not exist | New Diffuse midline glioma H3 K27M-mutant |
| Gliomatosis cerebri | Deleted |
| Protoplasmic astrocytoma Fibrillary astrocytoma | Deleted |
NOS: Not otherwise specified
IDH is a cytosolic enzyme that catalyzes the oxidative decarboxylation of isocitrate into alpha-ketoglutarate and nicotinamide adenine dinucleotide phosphate (NADPH) in normal cells9. The most common mutation involves amino acid 132 of IDH1 (R132H) in more than 70% of WHO grade II and III astrocytomas, oligodendrogliomas, and in secondary glioblastomas. As a surrogate to molecular genetic testing, a mutation-specific antibody can be used clinically to identify R132H mutations in glioma tumor tissue by immunohistochemistry (Figure 1). IDH2 (functions in the mitochondria) mutations noted in R172 amino acid are much less common (~3%) and associated with oligodendroglial histology2,10. The mutated IDH enzymes convert isocitrate to 2-hydroxyglutarate, which is believed to function as an oncometabolite and cause tumorigenesis. Although the exact mechanisms of this process remain to be elucidated, epigenetic mechanisms causing development of hypermethylated phenotype, thereby impairing differentiation has been hypothesized11,12. Retrospective analyses of tissues from multiple clinical trials have indicated IDH mutation to be a favorable prognostic marker in adult low and high grade gliomas10,13,14. The long-term data of RTOG 9802 trial3, which included high risk grade II glioma patients, reported outcomes stratified by IDH molecular status. Patients with tumoral IDH1 R132H mutations had significantly longer progression-free survival than did those without the mutation (p=0.003) and among those with the IDH1 R132H mutation, patients receiving procarbazine, CCNU and vincristine plus radiation did better than those receiving radiation therapy alone. EORTC 22033 also released their short-term follow up data in similar high risk grade II glioma patients15. This is an open label, randomized, phase III study to receive conformal radiation therapy versus dose-dense temozolomide 75mg/m2 daily for 21 out of 28 days repeated for 12 cycles. The patients were stratified by IDH and 1p/19q-codeletion status prior to randomization, making this a first-of-its-kind prospective study utilizing molecular markers in low-grade glioma. The results showed an improvement in progression-free survival (PFS) in patients with IDH-mutant and 1p/19q-intact tumors treated with radiation therapy than those treated with temozolomide and no differences in PFS in the IDH-mutant/1p/19q-codeleted and IDH-wildtype tumors. Median overall survival was not reached, and future data may help understand the predictive effect on different molecular subtypes of the two treatments. An orally available inhibitor of both IDH1/2 mutations is currently being tested in clinical trials in gliomas, acute myeloid leukemias and other solid tumors such as chondrosarcomas, and cholangiocarcinomas (NCT02746081, NCT02632708, NCT02987010 and NCT02577406). Inhibition of this enzyme is proposed to block the production of 2-hydroxyglutarate (2-HG) thereby impairing proliferation and promoting differentiation. Another potential target being explored is inhibition of glutamine synthesis, which in turn inhibits the conversion of glutamine to alpha keto glutarate16. Non-invasive metabolic imaging such as magnetic resonance (MR) spectroscopy can detect 2- hydroxy glutarate (HG) oncometabolite in tumors and help monitor treatment response17, but in some early studies such imaging has shown to be associated with false-negative results18,19.
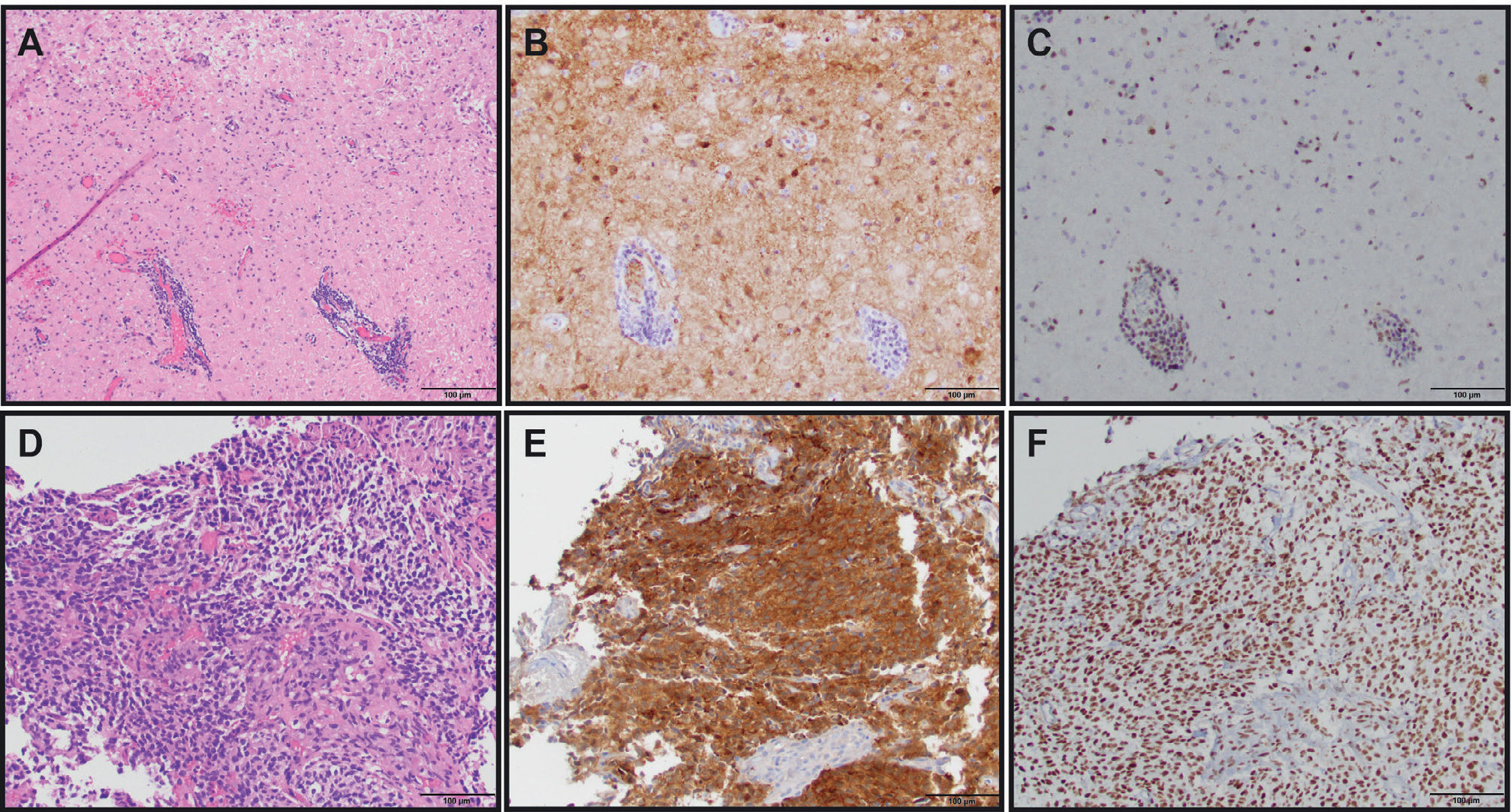
 Diffuse astrocytoma (gemistocytic variant), WHO grade II (H&E, 200X original magnification). (B) IDH1 R132H immunostain showing tumor cell positivity (IDH1 R132H, 200X original magnification). (C) ATRX immunostain showing endothelial cell and lymphocyte positivity as internal positive controls with tumor cells being negative (ATRX, 200X original magnification). Overall, the IDH and ATRX immunostains confirm the integrated diagnosis for images from A-C of diffuse astrocytoma, IDH-mutant with a likely concomitant ATRX inactivating mutation. (D) Anaplastic oligodendroglioma, WHO grade III with vascular proliferation (H&E, 200X original magnification). (E) IDH1 R132H immunostain showing tumor cell positivity (IDH1 R132H, 200X original magnification). (F) ATRX immunostain showing diffuse tumor cell positivity along with endothelial cell positivity as an internal positive control. In contrast to the astrocytoma, this oligodendroglioma did not show immunohistochemical evidence of an ATRX mutation; however, 1p/19q status by FISH confirmed the presence of 1p/19q-codeletion (image not shown). Therefore, the integrated diagnosis for images from D-F is anaplastic oligodendroglioma, IDH-mutant, 1p/19q-codeleted.")
THE USE OF MOLECULAR MARKER IMMUNOHISTOCHEMISTRY TO DIAGNOSE GLIOMAS
(A) Diffuse astrocytoma (gemistocytic variant), WHO grade II (H&E, 200X original magnification). (B) IDH1 R132H immunostain showing tumor cell positivity (IDH1 R132H, 200X original magnification). (C) ATRX immunostain showing endothelial cell and lymphocyte positivity as internal positive controls with tumor cells being negative (ATRX, 200X original magnification). Overall, the IDH and ATRX immunostains confirm the integrated diagnosis for images from A-C of diffuse astrocytoma, IDH-mutant with a likely concomitant ATRX inactivating mutation. (D) Anaplastic oligodendroglioma, WHO grade III with vascular proliferation (H&E, 200X original magnification). (E) IDH1 R132H immunostain showing tumor cell positivity (IDH1 R132H, 200X original magnification). (F) ATRX immunostain showing diffuse tumor cell positivity along with endothelial cell positivity as an internal positive control. In contrast to the astrocytoma, this oligodendroglioma did not show immunohistochemical evidence of an ATRX mutation; however, 1p/19q status by FISH confirmed the presence of 1p/19q-codeletion (image not shown). Therefore, the integrated diagnosis for images from D-F is anaplastic oligodendroglioma, IDH-mutant, 1p/19q-codeleted.
Combined loss of the short arm of chromosome 1 and the long of chromosome 19 is a hallmark genetic feature of oligodendrogliomas20. 1p and 19q codeleted tumors carry a much better prognosis in comparison to similar grade tumors without codeletion. A single deletion of either 1p or 19q does not carry the same prognostic significance and may in fact represent poorer prognosis. Histological evidence of an oligodendroglioma is almost always coexistent with IDH1/2 mutations21. Cairncross et al. in 1998 reported 1p/19q-codeletion status to be a predictive biomarker and prognostic with longer overall survival in anaplastic oligodendroglioma patients22. RTOG 940223 (neoadjuvant chemotherapy) and EORTC 2695124 (post–radiation chemotherapy) both utilizing combination chemotherapy of procarbazine, CCNU and vincristine showed doubling of survival in patients treated with initial chemoradiation versus radiation therapy alone, thereby confirming the predictive role of this molecular marker. RTOG 9802 (3) using a similar approach in high-risk grade II tumors did not have a sufficiently large enough sample size to measure the differential effect, although the European study EORTC 22033 did report best outcomes in IDH-mutant and 1p/19q-codeleted tumors15. Less responsiveness of the IDH-mutant non-codeleted tumor patients to chemotherapy alone highly suggests the predictive role of this biomarker. 1p/19q-codeletion testing can be done either by fluorescent in-situ hybridization, polymerase chain reaction (PCR)-based microsatellite analysis, or the use of newer methods such as microarrays, all of which require sufficient time and are usually not available at the time of diagnosis. A newer biomarker, mutated ATRX, is almost always mutually exclusive of 1p/19q-codeletion25 and can be interrogated quickly through immunohistochemistry testing (IHC loss indicating presence of the inactivating mutation). This testing is increasingly becoming part of the diagnostic algorithm, as indicated in the updated 2016 WHO classification (Figure 1).
MGMT PROMOTER METHYLATIONEpigenetic silencing of the MGMT (O6-methylguanine–DNA methyltransferase) gene by promoter methylation has been associated with longer overall survival in patients with newly diagnosed glioblastoma treated with alkylator chemotherapy, especially in the elderly population26,27, which resulted in MGMT testing as standard of care in this patient population. In the exploratory analysis of patients with anaplastic astrocytoma in the NOA-4 trial, MGMT promoter methylation predicted benefit to alkylator chemotherapy only in patients with IDH-wildtype but not in IDH-mutant tumors14. This will need further testing in prospective clinical trials. The technique of testing MGMT in tumor specimens has been an issue of ongoing debate. In clinical practice DNA-based methylation-specific PCR (MS-PCR) is the most commonly used test. MGMT protein testing by IHC, real time PCR, methylation specific pyrosequencing, methylation-specific multiplex ligation–dependent probe amplification, and mRNA expression testing are some of the other MGMT analysis methods. There is currently a lack of consensus regarding the most optimal method and the cut-off values used for each testing, which makes interpretation of clinical studies difficult. Another challenge is the heterogeneity of tumor specimens, especially when biopsies alone are available which results in variable frequency of methylation thereby making the final determination of methylation difficult. Approximately 15% of patients treated with temozolomide survived 2 years or more despite having MGMT nonmethylated tumors28. This suggests other mechanisms that may be responsible for improved response to treatment. One such mechanism that has been proposed is the presence of the T allele rs16906252 T genotype, which has been shown to be associated with better survival, irrespective of tumor methylation status29. Although a large majority of recurrent GBM tumors have shown to have retained the MGMT methylation status, their preferential response to chemotherapy in this setting has not been statistically proven30–33. MGMT methylated status has been significantly correlated (30%) with the incidence of pseudoprogression34 which is believed to reflect increased glioma killing effects of treatment. It is usually seen within the first 3 months of chemoradiotherapy treatment, but may be seen up to 6 months making interpretation of followup MRI scans complicated.
EGFR MUTATIONEpidermal growth factor receptor (EGFR) is a 170-kDa transmembrane glycoprotein with an extracellular ligand binding domain and a cytoplasmic domain containing a tyrosine kinase. It is associated with a more aggressive phenotype and is overexpressed in the small cell variant of glioblastoma35,36. Although preclinical results suggested activity of monotherapy with tyrosine kinase inhibitors37, the clinical trials using this mechanism failed to show therapeutic activity38 suggesting that alternative kinase signaling pathways may be involved or there is heterogeneous expression of EGFR39. An in-frame deletion of 801 base pairs in the extracellular domain of EGFR gene defines EGFRvIII, a genetic variant of the EGFR gene that is associated with poor survival40, but this has been debated in some other studies41. EGFRvIII has been reported in about 50% of EGFR-amplified glioblastomas and in 20–30% of primary glioblastoma42. A targeted peptide vaccine against EGFRvIII conjugated with keyhole limpet hemocyanin (KLH) paved the way for exploring immunotherapy as a treatment option in malignant gliomas43. In a Phase II study (ReACT)44, bevacizumab-naïve patients in their first or second relapse with EGFRvIII-expressing GBM were randomized in to bevacizumab plus either rindopepimut or KLH. As per the last update 25% of patients treated with rindopepimut plus bevacizumab remained alive at 2-years compared with none in the control arm. The median overall survival (OS) with rindopepimut was 11.3 versus 9.3 months in the control arm [hazard ratio (HR), 0.53; 95% confidence interval (CI), 0.32–0.88; P=0.0137]. Full response, PFS, and OS data are awaited. However in the recently reported results using the same peptide vaccine when added to standard of care as first line therapy in patients with tumors expressing EGFRvIII in a Phase III trial, it failed to reach its OS endpoint45. The future of using this immunotherapeutic option perhaps lies more in the recurrent setting when added to antiangiogenic therapy as indicated by some preclinical data46.
ATRX MUTATIONThe alpha thalassemia/mental retardation syndrome X linked (ATRX) gene mutations in glioma is primarily seen in in adolescents and young adults. Mutations in ATRX result in loss of ATRX protein by immunostaining and are thought to mediate loss of function. These inactivating mutations are known to correlate with the alternating lengthening of telomeres (ALT) phenotype and are associated with telomere dysfunction and other mutations including IDH1 and TP53, but are mutually exclusive from 1p/19q-codeletion47,48. In a large study ALT phenotype was associated with loss of ATRX protein expression in both pediatric and adult astrocytomas, suggesting ATRX loss to be a highly specific biomarker of astrocytic lineage49. This has now been incorporated in the decision making algorithm for differentiating oligodendroglial versus astrocytic origin of gliomas in the 2016 WHO classification. Given the ease of detection of this mutation by using immunohistochemistry, it makes it more accessible and feasible in daily practice. Within the subgroup of IDH-mutant astrocytic tumors, ATRX loss indicates a better prognosis as shown in some studies, perhaps due to the glioma-CpG island methylated phenotype (G-CIMP phenotype) that they represent50.
BRAF FUSIONS AND MUTATIONSBRAF is a member of RAS/RAF/MEK/ERK protein kinase pathway. It plays a key regulatory role in cellular proliferation and cell survival51. The most widely known BRAF mutation was initially reported in melanomas as point mutation (BRAFV600E), but it has now been recognized of importance in papillary thyroid cancer, colorectal cancer, hairy cell leukemia, and in gliomas. BRAF alterations are found in approximately 85% of pediatric low grade gliomas52. KIAA1549-BRAF fusion has been reported in 59-90% pilocytic astrocytomas (PAs) especially in the posterior fossa and is now increasingly being utilized as a diagnostic marker for PAs where pathological diagnosis is difficult53,54. Other fusion partners with BRAF have also been described, but are much less commonly seen. Point mutation in BRAF V600E has been reported in up to 80% cases of pleomorphic xanthoastrocytoma and 20% of gangliogliomas55,56. Some pediatric diffuse astrocytomas may also harbor BRAFV600E mutations57. Targeted molecular therapies affecting MAPK pathway have revolutionized the treatment of melanoma. Similar therapies are now being investigated in pediatric clinical trials58.
HISTONE H3 K27M MUTATIONA new group of mutations involving histones was recently described in high grade gliomas. Initially found in diffuse intrinsic pontine gliomas, these mutations have now been identified in both pediatric and adult gliomas59. HIST1H3B and H3F3A are the genes of interest that both encode histone H3 protein variants: H3.1 and H3.3, respectively. The two common mutations identified show preference for tumor location with K27M-mutants often seen in midline tumors (thalamus, pons, and spinal cord) and G34R/V-mutants seen in hemispheric tumors. The K27M mutation leads to altered post-translational modification of histone H3 causing impaired DNA methylation that is thought to drive gliomagenesis60–62.
With the discovery of this group of mutations, the updated WHO Classification of Tumours of the Central Nervous System included a new entity named “diffuse midline glioma, H3 K27M-mutant.” This follows suite with the new trend of an integrated histologic and molecular diagnosis as this tumor can only be diagnosed in the presence of a K27M mutation63. In fact, a recent series of 47 infiltrative gliomas found H3 K27 mutations in multiple midline locations in both children and adults, but all with varying histologic appearances. Tumors ranged from classic lower grade infiltrating astrocytomas to glioblastomas (GBMs) with variants including giant cell GBM, epithelioid GBM, rhabdoid GBM, GBM with PNET-like foci, and gliosarcoma. One tumor was even histologically classified as a pilomyxoid astrocytoma64.
The wide variation in histologies found to harbor H3 K27M mutations makes it imperative for us to have the ability to test for this mutation in pediatric and adult gliomas. A mutant-specific antibody that detects K27M mutations in both H3.1 and H3.3 is now widely accepted as a surrogate for molecular testing. Many groups have published using this antibody and suggest liberal application for all midline gliomas61,63. Not only is this testing important for proper classification of gliomas, but also prognosis as patients with H3 K27M-mutated tumors do worse65. And for near-future use, targeted histone-modifying enzymes are under investigation for potential therapy in patients with known histone H3 mutations with great success in pre-clinical models66.
DISCUSSIONMolecular makers in gliomas have improved our understanding of mechanisms of gliomagenesis and helped define various molecular and histologically integrated diagnoses; however, they have also raised several challenges that we have begun to encounter during clinical care of these patients.
Prognostic valueCounselling patients regarding prognosis may become a challenge in specific situations such as IDH wildtype grade II and III gliomas, which are increasingly being recognized as poor prognostic category even when compared with IDH mutant glioblastoma2,10. The importance of grade as a prognostic marker is perhaps being lost, which is necessary, but complicated to incorporate during conversations with our patients. For example, using The Cancer Genome Atlas data, stratification of clinical risk was better defined using molecular markers for IDH-mutant, 1p/19q-codeleted tumors (oligodendrogliomas)48.
Clinical ManagementMost of the current available molecular data has been obtained from retrospective evaluation of tissue. Some of the larger datasets did not include patients receiving similar regimens of treatment. This makes the interpretation of impact of treatment with the given molecular biomarkers increasingly difficult. It is unclear from the datasets if standard radiation therapy and or chemotherapy were used for patients during the study time frame. Combination chemoradiotherapy has now emerged to be superior to radiation therapy alone with respect to survival in almost all histologies of gliomas3,67,68 however datasets such as TCGA did not include treatments for survival analysis which is the key aspect that remains unaccounted for. How our choice of treatment is influenced by the presence of specific biomarkers will need to be prospectively validated in clinical trials.
Predictive value1p/19q codeletion had a well-defined role as a predictive biomarker for chemotherapeutic response but with the new classification has primarily emerged as a diagnostic marker for oligodendroglioma histology. Recent trial has confirmed the predictive value of MGMT promoter methylation status in response to temozolomide chemotherapy especially in the elderly patients with glioblastoma27,69. Ongoing trials targeting IDH1/2 mutations in glioma (NCT02481154) if proven to have efficacy may then make these mutations a predictive biomarker in addition to their diagnostic and prognostic value. Epithelioid GBMs have shown higher incidence of BRAFV600E mutations70. There have been several case reports showing sustained responses and even regression of tumors with the use of BRAF targeted therapy in gliomas with BRAFV600E mutation71–74. Therapeutic value of BRAF inhibitors and combination of BRAF and MEK inhibitors which have shown greater promise in melanoma studies75 will need to be evaluated in larger glioma trials. Immune checkpoint inhibitors are showing promising activity in various solid and hematological cancers. Expression of programmed cell death 1 (PD1) on immune cells and programmed cell death ligand 1 (PDL1) on tumor cells have been proposed to have predictive value for responses to immune checkpoint inhibitors. In glioblastomas, PD1 and or PDL1 expression has been reported in majority of the cases and PDL1 expression is associated with worse outcomes76,77. Clinical trial outcomes utilizing immune checkpoint blockers, anti-programmed cell death 1 and anti-PDL1 in glioblastoma are desperately awaited. If shown to have response then PD1/PDL1 markers may gain a predictive value in gliomas.
Clinical trial design and conductClinical trial designs in glioma are increasingly utilizing molecular data in stratifying patients. Molecular testing is almost entirely dependent on the availability of adequate tissue. Neurosurgeons are constantly faced with technical challenges when attempting to obtain tissue especially when tumor is in eloquent areas. Hence the tissue obtained from surgical resection or biopsy could be scant, which may be critical in terms of molecular testing for individual trials for eligibility. This suggests an increasing need for universally accepted standardized testing for molecular testing such as MGMT promoter methylation for trial eligibility. Appropriate use of historical controls for intervention comparison poses another challenge since the specific treatment regimens of patients included in large datasets are not known. Moving forward, concurrent control studies will require larger patient populations to accrue and long followup for IDH-mutant or 1p/19q-codeleted arms. The question of right surrogate for efficacy of newer drugs with the knowledge of these biomarkers will need to be addressed as well. With the survival of some patient populations now averaging several years (RTOG 9802) the survival endpoint would mean longer follow up for data to mature. Imaging as an endpoint, specifically in the case of IDH-mutant tumors measuring 2HG metabolite by MR spectroscopy, appears to be promising. Neurocognitive evaluation will also need to be incorporated routinely in clinical trials in patients with good prognostic markers.
CONCLUSIONMolecular markers are now incorporated as diagnostic and prognostic indicators in the updated WHO 2016 classification. Newer entities have been defined and some removed in comparison to the WHO 2007 classification. Many markers have increased our understanding of gliomagenesis and newer targeted drugs showing promise in preclinical studies are being evaluated in clinical trials. Challenges in clinical practice and clinical trial designs are increasingly being recognized and will need to be addressed with future studies.
The authors declare no conflicts of interest, in relation to this article.