


Colorectal cancer (CRC) is one of the most common solid tumors worldwide. Consumption of dietary fiber is associated with a low risk of developing CRC. The fermentation of the dietary fiber by intestinal microflora results in production of butyrate (BT). This short-chain fatty acid is an important metabolic substrate in normal colonic epithelial cells and has important homeostatic functions at the colonic level. Because the cellular effects of BT (e.g. inhibition of histone deacetylases) are dependent on its intracellular concentration, knowledge on the mechanisms involved in BT membrane transport and its regulation seems particularly relevant. In this review, we will present the carrier-mediated mechanisms involved in BT membrane transport at the colonic epithelial level and their regulation, with an emphasis on CRC. Several xenobiotics known to modulate the risk for developing CRC are able to interfere with BT transport at the intestinal level. Thus, interference with BT transport certainly contributes to the anticarcinogenic or procarcinogenic effect of these compounds and these compounds may interfere with the anticarcinogenic effect of BT. Finally, we suggest that differences in BT transport between normal colonocytes and tumoral cells contribute to the “BT paradox” (the apparent opposing effect of BT in CRC cells and normal colonocytes).
Cancer is the second leading cause of death, after cardiovascular diseases, in occidental countries1. Colorectal cancer (CRC) is one of the most common malignancies and cause of cancer death in developed countries,2 including United States3 and Europe.4 In Europe, CRC is the third most common type of cancer in both men and women and the second leading cause of cancer related-death.2 The prevalence of CRC has been steadily increasing over the last century, possibly as a result of industrialization and changes in life style/environmental/dietary factors.2
Both epidemiological and experimental animal studies have shown that dietary fiber possesses a protective role in CRC.5,6 Dietary fiber exerts a protective role against CRC through a variety of mechanisms, including reduced concentrations of intestinal carcinogens owing to increased stool mass, decreased transit time, and bacterial fermentation of resistant starch to short-chain fatty acids (SCFAs: acetate, propionate and butyrate) in the colon.6,7 Among SCFA, butyrate (BT) plays a key role in colonic epithelium homeostasis, by having multiple regulatory roles at that level, namely: (1) it is the main energy source for colonocytes; (2) it promotes growth and proliferation of normal colonic epithelial cells; (3) it inhibits colon carcinogenesis; (4) it inhibits colon inflammation and oxidative stress; (5) it improves the colonic defense barrier function; (6) it stimulates fluid and electrolyte absorption; (7) it stimulates mucus secretion and increases vascular flow and motility; and (8) it reduces visceral perception, intestinal discomfort, and pain.8,9
Several lines of evidence support an important role of BT in the prevention/inhibition of colon carcinogenesis.10,11 In vitro, BT suppresses growth of cancer cells, inducing differentiation and apoptosis and inhibiting cell proliferation.12,13 Also, several well-designed animal models have demonstrated a protective effect of BT on colorectal carcinogenesis.14–19 Moreover, there is an inverse relationship between the levels of BT in the human colon and the incidence of CRC,20 and an increased incidence of tumors in the distal colon, where the concentration of BT is lower, suggesting an inverse relationship between BT and CRC.21
The molecular mechanism by which BT inhibits colon carcinogenesis seems to involve various effects on gene expression, which are mainly attributed to its capacity to act as an histone deacetylases (HDAC) inhibitor, leading to hyperacetylation of histones.22,23 HDACs can regulate the expression of a large number of genes by direct interaction with transcription factors such as p53, retinoblastoma protein, Stat3, NF-kB and estrogen receptors8 and histone deacetylase inhibitors (HDACi) are critical epigenetic regulators and a new class of anticancer agents.24 It is likely that BT has also some other intracellular targets, including DNA methylation,25 histone methylation,26 hyperacetylation of nonhistone proteins,27 inhibition of histone phosphorylation,28 regulation of expression of micro-RNAs (miRNA)29,30 and modulation of intracellular kinase signaling.31–33
More recently, BT was demonstrated to elicit cellular uptake-independent biologic effects on colonic epithelial cells.34 Indeed, BT was found to be a physiologic agonist of GPR109A, a G-protein-coupled receptor which is abundantly expressed in the apical membrane of mouse and human colonic epithelial cells.34,35 SCFAs were also reported to be GPR41 and GPR43 ligands.36–38 GPR109A and GPR43 appear to act as a colonic tumor suppressors and also to be involved in the anti-inflammatory effect of SCFA.35,36,39,40
Intestinal transport of BTThe most important molecular mechanisms involved in the anticarcinogenic effect of BT are dependent on its intracellular concentration (because HDAC expression is overregulated,41,42 while BT membrane receptors (GPR109A and GPR43) are silenced or downregulated in CRC34,38). So, knowledge on the mechanisms involved in its membrane transport is relevant to both its physiological and pharmacological benefits. Also, changes in transporter expression or function will have an obvious impact on the effect of BT, and therefore, knowledge on the regulation of its membrane transport seems particularly important.
BT is a weak acid (pKa=4.8) and more than 90% exist in the ionized form under physiological conditions in the colon (pH 5.5–6.7), thus requiring a transporter for absorption.43,44 BT is preferentially absorbed in the proximal part of colon where the highest luminal concentration occurs.45–47 Several different mechanisms for BT uptake across the apical membrane of colonocytes have been proposed, including simple diffusion of the undissociated form (in the distal colon),43,44 counter-transport with bicarbonate (BT/HCO3− exchanger)48,49 and transport by monocarboxylate transporters.50,51 The two major monocarboxylate transporters identified for BT absorption across the luminal membranes of colonocytes are the proton-coupled monocarboxylate transporter 1 (MCT1) and the sodium-coupled monocarboxylate transporter 1 (SMCT1).50–52 These will be next described.
Monocarboxylate transporter 1 (MCT1)The monocarboxylate transporter (MCT) family is composed by 14 members encoded by the SLC16 gene family.53 The MCT1 (SLC16A1) gene was cloned in 199454 and the structural gene organization as well as isolation and characterization of the SLC16A1 promoter were later described.55 MCT1 is composed by 500 amino acids, is well conserved and is ubiquitously expressed in almost every tissue.56 MCT1 protein levels vary along the human digestive tract: the expression of MCT1 is very low in the small intestine but it increases in the colon with maximal levels in the distal segment, being confined to the upper regions of colonic crypts.57,58 The precise subcellular localization of MCT1 remains controversial: a predominant basolateral57,59,60 or apical membrane localization58,61 or its presence in both cell membranes62 has been described.
MCT1 translocates a proton through the plasma membrane together with a molecule of BT.63 Physiologically, MCT1 is probably more active in the proximal colon, because its Km for BT is about 2.4–2.8mM, and there is a higher concentration of BT (in the mM range after digestion of dietary fiber) and the luminal pH is lower in this region.21
MCT1 transports a variety of natural substrates including monocarboxylates (e.g. BT) and ketoacids.53 Moreover, numerous drugs containing a carboxyl group in their chemical structure and/or weak organic acids may be potential substrates for MCT1. Examples of such drugs are salicylic acid, nonsteroidal anti-inflammatory drugs (NSAIDS), benzoic acid, nicotinic acid, lonidamine, cholesterol synthesis inhibitors and some β-lactam antibiotics.64–66
MCT1 regulationTransporter regulation includes transcriptional (activators and repressors), post-transcriptional (splice variants), chromosomal (epigenetic modifications), translational (mRNA stability) and post-translational (alteration of protein) modifications. MCT1 is known to be regulated at the transcriptional and post-transcriptional level, and at the level of transporter activity.
MCT1 is known to be regulated at various points during gene expression (transcriptional regulation). SLC16A1 promoter has putative binding site sequences for the transcription factors upstream stimulatory factor (USF) 1 and 2 (MCT1 repressors),67 NF-κB (involved in BT-induced MCT1 upregulation),68 activated protein 1 and 2 (AP1 and AP2) and stimulating protein-1 (Sp1).55 Also, the co-activators peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α)69 and peroxisome proliferator-activated receptor alpha (PPARα)70,71 upregulate MCT1. Hormone regulation has also been described. Somatostatin,72 leptin,73 thyroid-stimulating hormone74 and testosterone75 induce MCT1 expression.
MCT1 is also under post-transcriptional regulation by microRNAs. The MCT1 untranslated region is a target for three microRNAs (miR-29a, miR-29b, and miR-124), and recently, miR-29a and miR-29b have been described to silence MCT1 expression.76
MCT1 activity is modulated by several compounds, described as classic inhibitors of MCT1: (1) bulky or aromatic monocarboxylates like α-cyano-4-hydroxycinnamate; (2) inhibitors of anion transport such as 5-nitro-2-(3-phenylpropylamino)benzoate; (3) thiol reagents, such as p-chloromercuribenzene sulphonate (pCMBS), and amino reagents.53
MCT1 expression is downregulated in the inflamed mucosa of inflammatory bowel disease patients, in animal models of inflammation, and in response to the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ).77 Interestingly, infliximab (an anti-TNF-α monoclonal antibody) markedly increased MCT1 mRNA levels in the inflamed colon of Crohn's disease patients.57 The reduction in the cellular uptake of BT occurring during inflammation may contribute to the fact that inflammatory bowel disease is associated with an elevated risk of CRC.77,78
Physical activity, known to reduce CRC risk,79 increases MCT1 protein expression and activity in muscle.80,81 Because MCT1 is substrate-induced by lactate,82 the increased blood lactate levels found after physical activity can affect MCT1 levels in colon.
MCT1 is associated with a plasma accessory protein, the membrane glycoprotein CD147 (also known as basigin, EMMPRIN, OX-47 or HT7), and this ancillary protein is known to be involved in regulating MCT1 localization and activity.83
MCT1 is also functionally coupled to proteins involved in acid/base regulation. Indeed, carbonic anhydrase,84 Na+/HCO3− cotransporter and Na+/H+ exchangers85 enhance MCT1 transport activity.
Several nutrients and xenobiotics present in the diet were also found to affect MCT1 expression and activity. Intestinal MCT1 gene expression is decreased by the polyphenols EGCG, myricetin, and catechin,12 by caffeine, tetrahydrocannabinol and MDMA (ecstasy),86 by high-protein diets (linked to NH3- and TNF-α-mediated signaling)87 and by fasting.88 In contrast it is increased by chrysin.86 The expression of MCT1 as well as its abundance in the apical surface of the colonic mucosal sections increased in pectin-fed rats compared with rats on fiber-free diet.62,89 Moreover, substrates of MCT1 such as BT increased MCT1 protein expression in colon68,90,91 and its apical localization.92 Recently, substrate (e.g. BT)-induced enhancement of MCT1 surface expression and function was found to be mediated by a novel nutrient sensing mechanism involving GPR109A as a SCFA sensor.89 MCT1 activity is also modulated by several exogenous compounds. Acutely, MCT1 is inhibited by NSAIDs (e.g. acetylsalicylic acid and indomethacin),86,93,94 some phytochemicals (e.g. resveratrol, quercetin, myricetin, chrysin, (−)-epicatechin and (−)-epigallocatechin gallate),12,94–96 xanthines (caffeine and theophylline) and acetaldehyde.86 Chronically, MCT1 activity is inhibited by tetrahydrocannabinol, MDMA,86 the bile salt chenodeoxycholic acid,97 enteropathogenic Escherichia coli,98 IFN-γ and TNF-α77 and is increased by caffeine (an effect not related to changes in the expression level of MCT1, as this agent decreased this parameter; see above),86 some phytochemicals (e.g. quercetin, EGCG, rutin, chrysin, myricetin and catechin)12 and some mineral waters (Melgaço® and Vidago®).99 Of note, Lactobacillus acidophilus counteracts E. coli-induced inhibition of BT uptake in intestinal epithelial cells.100 In conclusion, numerous nutrients and xenobiotics can modulate MCT1 expression and activity, and competition for the same transport pathway between BT and these compounds can cause a significant change in BT absorption.
MCT1 and CRCThe first report on MCT1 protein expression in human tumor samples described a decrease in MCT1 expression in colonic transition from normality to malignancy.61,101 Evidence for MCT1 downregulation was later observed also in other cancer types.61,102 The loss or silencing of MCT1 has been demonstrated to correlate with: (a) transition from normality to malignancy in colonic epithelium,61 (b) dysregulation of BT-responsive genes involved in differentiation and apoptosis101,103 and (c) an important metabolic switch from BT β-oxidation to glycolysis. In relation to this last point, it should be noted that BT is the main energy source for colonocytes, accounting to about 70% of total energy utilization.104 However, CRC cells show a reduction in BT uptake as a result of reduced MCT1 (and SMCT1; see below) expression,104,105 associated with an increase in the rate of glucose uptake (via an upregulation of facilitative glucose transporters (e.g. GLUT1))106 and glycolytic oxidation (via an increase in the expression levels and activity of glycolytic enzymes).104,107,108
Although MCT1 expression decreases during colonic transition from normality to malignancy, being downregulated in the early stages of carcinogenesis,61 a later upregulation of MCT1 in advanced metastatic CRC tumors has been described.109,110 Solid tumors are usually exposed to low oxygen environments. Interestingly enough, although MCT1 expression is not HIF-1α induced,111 CD147, the accessory protein that MCT1 requires in order to function, is upregulated by HIF-1α under hypoxic microenvironment.112 In advanced CRC tumors, cells are highly glycolytic and convert the majority of glucose into lactate and thus cells must efficiently export lactate, in order to maintain a permissive intracellular pH, high glycolytic rates and ATP levels.113 MCTs are bidirectional transporters114 and powerful regulators of intracellular pH by extruding lactate together with a proton.113 Accordingly, MCT1 inhibition decreases intracellular pH, resulting in tumoral cell death.108,115,116 So, upregulation of some MCT isoforms may occur as a means of exporting lactate. In this context, lactate transporters (MCTs) are currently seen as potential therapeutic targets in cancer treatment, with promising results having been obtained.117–119 However, systemic delivery of MCTs (and more specifically, MCT1) inhibitors could affect almost every organ of the body, with the most drastic effects on cardiac and skeletal muscle.120 Also, in CRC cells, both MCT1 and MCT4 appear to play a pivotal role in lactate transport and tumor survival and development.121,122 However, no specific MCT4 small molecule inhibitor has been identified so far.123 BT was previously approved for clinical use in CRC treatment,124 because BT is a substrate of both MCT1 and MCT4 and is well metabolized, having no side effects.120 Because BT competes with lactate for MCT1 and is a HDACi, it is a good compound to test in the context of inhibition of lactate release in CRC.125 So, although the anticarcinogenic effect of BT is believed to result from HDAC inhibition,23 it is also interesting to speculate that MCT-mediated BT uptake may result in a decrease in the intracellular pH, originating tumoral cell death.
Sodium-coupled monocarboxylate transporter 1 (SMCT1)SMCT1 is a member of the sodium solute symporter family (SLC5), first cloned in 2002.126 This transporter is encoded by the SLC5A8 gene127 and the SLC5A8 gene promoter has also been characterized.128 SLC5A8 encodes a protein with 610 amino acids,129 having a restricted distribution (primarily kidney and intestine).130 At the intestinal level, SLC5A8 is abundantly expressed in the apical membrane of the ileum and colon,131,132 and its levels are highest in distal colon, followed by proximal colon and ileum.78 SLC5A8 transports a variety of monocarboxylates such as lactate, pyruvate and γ-hydroxybutyrate (GHB),132,133 ketone bodies,134 nicotinate structural analogs,135 pyroglutamate (amino acid derivative)136 and benzoate and its derivatives (salicylate and 5-aminosalicylate).135 SLC5A8 has been characterized as a Na+-coupled BT transporter137,138 being thus also referred as sodium-coupled monocarboxylate transporter 1 (SMCT1).130 Recent studies showed that SMCT1 is also present in normal human intestinal cell lines78,94; however, no studies have characterized SMCT1 function in the native human intestine. Because SMCT1 has a low Km (50μM) for BT139 and is more expressed in distal colon,78 SMCT1 is probably physiologically more important in the distal colon (where the concentrations of BT are lower).21
SMCT1 regulationKnowledge on the regulation of SMCT1 at the intestinal level is very scarce. SMCT1 has been found to be inhibited by some NSAIDs (ibuprofen, ketoprofen, fenoprofen, naproxen135 and indomethacin94), phytochemicals (resveratrol and quercetin94), TNF-α,76 oxidative stress,140 chenodeoxycholic acid97 and by the absence of gut commensal bacteria.141 On the contrary, SMCT1 was found to be stimulated by some other NSAIDs (diclofenac, meclofenamate and sulindac142), by activin A143 and by the probiotic Lactobacillus plantarum.76
Obesity and diabetes are risk factors for developing CRC143,144 and it is interesting to verify that ob/ob mice (an animal model of obesity) show a decrease in SMCT1 protein intestinal levels.145 Inflammatory bowel disease is also associated with an increased risk for CRC,9 and SMCT1 is also downregulated during inflammation.78
SMCT1 and CRCStudies have shown that SMCT1 expression is frequently silenced in aberrant crypt foci (the earliest detectable morphologic abnormality of the colonic epithelium), colon adenomas, colon cancers and colon cancer cell lines, suggesting that SMCT1 silencing is an early event in colon tumorigenesis.127,130 Interestingly, CRC patients often have allelic loss of chromosome 12q, which contains the SLC5A8 gene.146,147 SMCT1 is also silenced in cancers of the thyroid, head and neck, breast, stomach, prostate, pancreas and blood.130,148,149 SMCT1 is silenced by aberrant DNA hypermethylation.148,150 Therefore, SMCT1 was proposed to function as a tumor suppressor, the ability of this transporter to mediate the entry of BT into colonocytes underlying its potential tumor suppressor function,52,127,130 and combination of upregulation of matrix metalloproteinases-7 and SMCT1 downregulation is an optimal biomarker for identifying CRC cases.151 Also, SMCT1 activity is positively correlated with CRC remission and patient survival.152 However, Smct1-null mice do not reveal a higher incidence of tumors in the colon under optimal dietary fiber conditions, possibly because BT is transported by Mct1 under these conditions. But, under low-fiber dietary conditions, the incidence of CRC is much higher in Smct1-null mice, possibly because, being the luminal concentrations of BT much lower, it is not transported by Mct1 and Smct1 becomes the most important BT transporter.153,154 Recently, ectopic expression of SMCT1 in cancer cells (that have SMCT1 silenced) was found to induce the translocation of the anti-apoptotic protein survivin to plasma membrane and cell cycle arrest, apoptosis and enhancement in chemosensitivity, independently of the transport function of SMCT1 and of the histone acetylation status of the cell.155 So, the tumor-suppressive role of SMCT1 does not depend exclusively on the ability of this transporter to mediate the entry of HDAC inhibitors such was BT and pyruvate into cells.
Breast cancer resistance protein (BCRP)BT cellular pools are not only dependent on the above BT uptake systems but also depend on efflux transporters, capable of removing BT from the cells. The ATP-binding cassette (ABC) transporter superfamily includes membrane proteins that translocate a wide variety of substrates across membranes.156 The human intestinal tract expresses high levels of some ABC transporters (e.g. P-glycoprotein (MDR1; encoded by ABCB1), multidrug resistance protein 1 (MRP1; encoded by ABCC1), and the breast cancer resistance protein (BCRP; encoded by ABCG2)), and these efflux transporters are believed to be involved in limiting drug absorption, bioavailability, and toxicity.156 We recently verified that BT is a BCRP substrate. Interestingly, inhibition of BCRP significantly potentiated the inhibitory effect of BT upon cell proliferation.157
BCRP and CRCBCRP mRNA and protein expression are significantly downregulated in human colorectal adenomas,156 in ApcMin mice (a mouse model of colon cancer),156 in human CRC tissue and in most human cancers,158 suggesting that malignant transformation of the colonic epithelium in vivo is accompanied by a significant downregulation of BCRP. Accordingly, we demonstrated that, contrary to non-tumoral intestinal epithelial cells (IEC-6 cells), Caco-2 cells (a tumoral cell line derived from a colon adenocarcinoma) do not show BCRP-mediated efflux of BT.157 BCRP expression is also dramatically reduced in inflammatory bowel disease,159–161 which is interesting in the context of the relationship between inflammatory bowel disease and CRC.156
The mechanisms involved in BCRP downregulation in CRC have not been much investigated. BCRP expression depends on Wnt/β-catenin signaling pathway in colon cancer cell lines162 and mutations of the β-catenin gene are frequently found in chemically-induced colon tumors in both rat and mouse carcinogenesis models163 and in human colon tumors,164 suggesting that inhibition of this signaling pathway may decrease BCRP expression. Studies also demonstrated that human BCRP polymorphisms causing a decrease in its activity could influence the individual susceptibility to cancer.165,166 Interestingly enough, BCRP exerts a protective function at the intestinal epithelial level by limiting the access of dietary mutagens and carcinogens.167,168 So, a decrease in BCRP expression/function may place an individual at greater risk of exposure to dietary/environmental carcinogens.
As mentioned above, CRC is associated with a downregulation of BCRP.158 However, some studies described BCRP overexpression in colorectal invasive cancers (i.e., advanced stage of carcinogenesis),169–171 suggesting that BCRP may be involved in cancer progression and metastasis, and some other studies showed that BCRP is overexpressed in solid tumors.172 Most solid tumors are exposed to low oxygen environments and BCRP gene transcription is activated by binding of HIF-1α to a hypoxia response element under low oxygen conditions.173 c-Myc overexpression also upregulates BCRP,174 and pregnane X receptor (also known as the steroid and xenobiotic sensing nuclear receptor) also contributes to upregulation of BCRP.175
An important point relates to the fact that BCRP is known to be overexpressed in cancer cell lines and tumors that are “multidrug resistant” (MDR).176 The term “multidrug resistance” (MDR) is used to describe the ability of cells exposed to a single drug to develop resistance to a broad range of structurally and functionally unrelated drugs.177 BCRP is one of the human ABC transporters implicated in MDR in cancer chemotherapy.178,179 BCRP recognizes and transports numerous anticancer drugs including conventional chemotherapeutic and relatively new molecules in clinical use: 5-fluorouracil, methotrexate, mitoxantrone, anthracyclines, daunorubicin, doxorubicin, topotecan, diflomotecan, irinotecan, tyrosine kinase inhibitors (e.g. imatinib and gefitinib) and nucleoside analogs.180–182 Thus, downregulation of BCRP expression and/or function has been proposed as part of a regimen to improve cancer therapeutic efficacy. Several specific inhibitors of BCRP have been reported, and some are currently undergoing clinical trials or are available to treat patients.172 BT, by competing with anticancer agents for BCRP, may increase their intracellular level, thereby increasing their cytotoxicity. In agreement with this, a synergistic effect of the combination of BT with anticancer agents transported by BCRP has been reported.180–182 So, interaction of BT with BCRP and with other BCRP substrates/inhibitors will have clinical implications for CRC therapy.180–183
BT transport and CRCBT is the main energy source for colonocytes and promotes growth and proliferation of normal colonic epithelial cells.184,185 However, BT suppresses the growth of cancer cells (inducing differentiation and apoptosis and inhibiting cell proliferation).13 This apparent opposing effect of BT upon growth of normal versus tumoral colonocytes has been referred to as the “BT paradox”.186
Differences in BT metabolism between normal and tumoral cells lines, more specifically the Warburg effect, may contribute to this “paradox”.186 In normal colonocytes, BT stimulates cell growth by functioning as an oxidative energy source; it is the primary fuel of these cells, being metabolized by β-oxidation followed by the tricarboxylic acid (TCA) cycle.102,187 On the contrary, BT is inefficiently metabolized in cancerous colonocytes, since glucose takes the place of BT as the major energy source (the Warburg effect), and it accumulates at greater levels inside of nuclei (being therefore a candidate oncometabolite), with a corresponding increase in HDAC inhibition.179,186 This results in inhibition of cell growth (Fig. 1). Interestingly, several studies have shown that high concentrations (5mM) of BT induces H3ac acetylation, decreases cell proliferation and viability and induces cell differentiation also in non-tumoral intestinal epithelial cell lines.157,179,186,188 This can be explained by the fact that 1–2mM corresponds to the oxidative capacity of these cells.189 Therefore, at concentrations greater than 2mM, BT accumulates and functions as a HDAC inhibitor also in normal colonocytes. Altogether, these findings lead to a model whereby BT facilitates the normal turnover of the colonic epithelium by promoting colonocyte proliferation in the bottom half of each crypt, where there is a low concentration of BT, while increasing apoptosis in those cells that exfoliate into the lumen (because of a higher concentration of BT).179,186
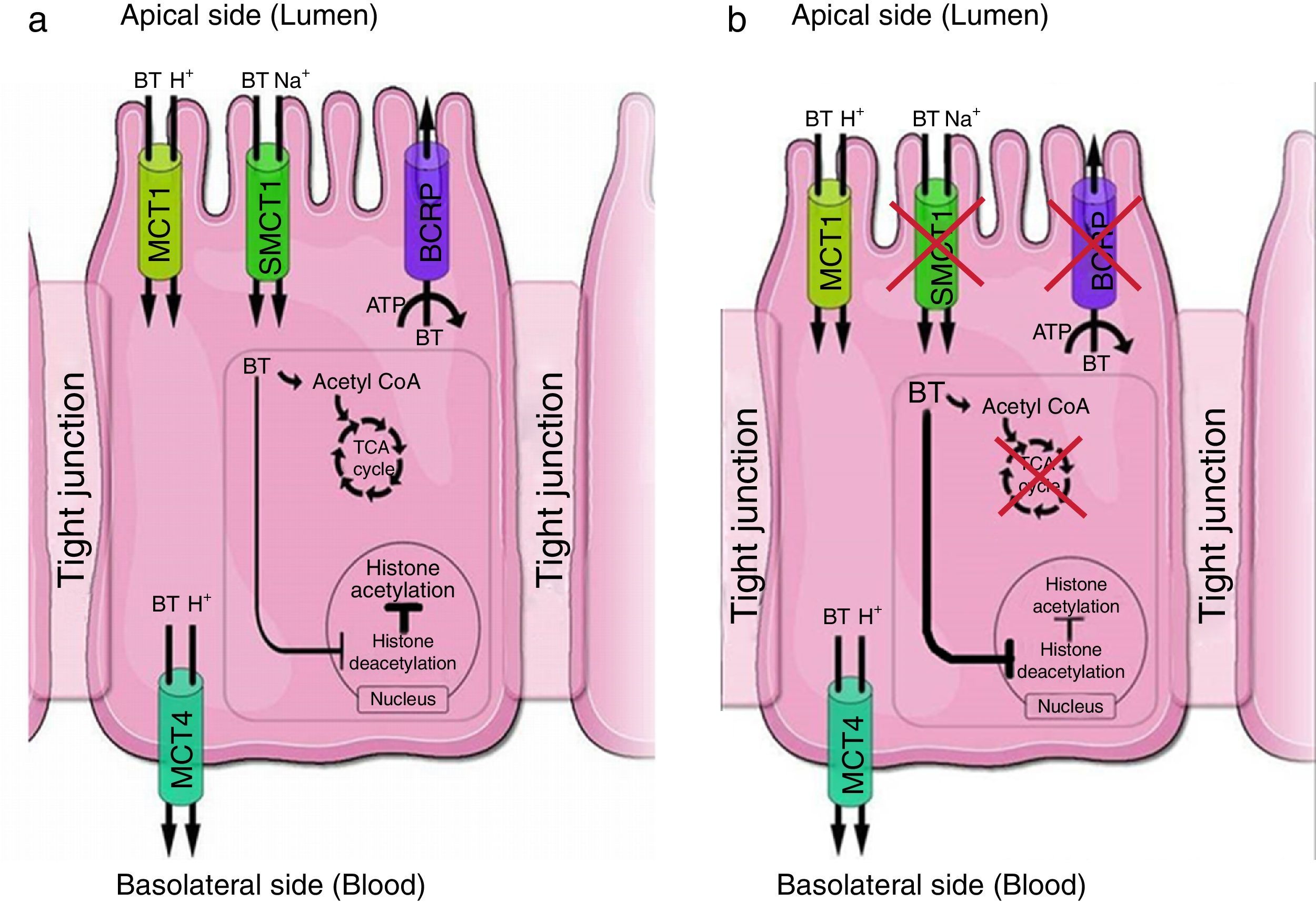
 Proposed model of expression and function of BT transporters in normal colonocytes. Transporters such as MCT1 (monocarboxylate transporter 1, gene name SLC16A1) and SMCT1 (sodium-coupled monocarboxylate transporter 1, gene name SLC5A8) mediate influx of BT at the apical membrane. BT is rapidly metabolized in the tricarboxylic acid (TCA) cycle. BCRP (gene name ABCG2), an ATP-dependent efflux transporter, mediates BT efflux at the apical membrane. At the basolateral membrane, efflux of BT occurs via MCT4 (monocarboxylate transporter 4, gene name SLC16A3). (b) Proposed model of expression and function of BT transporters in tumoral colonocytes. In tumoral colonocytes, glycolysis becomes the primary energy source, exceeding BT oxidative energetic metabolism, and cells rapidly convert the majority of glucose into lactate. BT, which is now less oxidized, behaves as a histone deacetylase inhibitor promoting histone acetylation. MCT1 mediates influx of BT and may also mediate efflux of lactate at the apical membrane. Adapted from Refs. 188,194.")
(a) Proposed model of expression and function of BT transporters in normal colonocytes. Transporters such as MCT1 (monocarboxylate transporter 1, gene name SLC16A1) and SMCT1 (sodium-coupled monocarboxylate transporter 1, gene name SLC5A8) mediate influx of BT at the apical membrane. BT is rapidly metabolized in the tricarboxylic acid (TCA) cycle. BCRP (gene name ABCG2), an ATP-dependent efflux transporter, mediates BT efflux at the apical membrane. At the basolateral membrane, efflux of BT occurs via MCT4 (monocarboxylate transporter 4, gene name SLC16A3). (b) Proposed model of expression and function of BT transporters in tumoral colonocytes. In tumoral colonocytes, glycolysis becomes the primary energy source, exceeding BT oxidative energetic metabolism, and cells rapidly convert the majority of glucose into lactate. BT, which is now less oxidized, behaves as a histone deacetylase inhibitor promoting histone acetylation. MCT1 mediates influx of BT and may also mediate efflux of lactate at the apical membrane. Adapted from Refs. 188,194.
Differences in HDACs expression levels between normal and tumoral cells lines also contribute to the “BT paradox”, because HDACs appear to be overexpressed in CRC cells.38,39 The Warburg effect appears to be a cause, rather than a consequence of this mechanistic shift in histone acetylation.179,186
Differences in BT transport between normal and tumoral cells lines also appear to contribute to the “BT paradox”. In normal colon epithelial cells, BT is taken up by MCT1 and SMCT1 located at the apical membrane52,94,103 and, being the main energy source for the colonocytes, it is metabolized.179,186 BT that is not metabolized is effluxed by BCRP, thus decreasing its intracellular concentration and so BT has no effect at HDACs157 (Fig. 1). This is consistent with the fact that in normal colonic tissue, BT does not inhibit cell proliferation.190–192 As shown above, studies described MCT1 upregulation in advanced metastatic CRC tumors. Therefore, in tumoral colon epithelial cells, BT is taken up by MCT1 located at the apical membrane86,103,109,110 but it is metabolized inefficiently due to the Warburg effect179,186; moreover, it does not suffer BCRP-mediated efflux.157 So, it accumulates at greater levels inside of nuclei179,186 thus acting as a HDAC inhibitor, leading to hyperacetylation of histones and to increased accessibility of transcription factors to DNA promoters,23 thus inducing apoptosis,12,13 inhibiting proliferation and promoting a more differentiated phenotype12,186,193 (Fig. 1).
ConclusionsBT is the main energy source for normal colonic epithelial cells and inhibits colon carcinogenesis. Because the cellular effects of BT (e.g. inhibition of histone deacetylases) are dependent on its intracellular concentration, knowledge on intestinal BT transport mechanisms and its regulation is crucial in the context of the physiological effects of BT and of CRC pathophysiology.
Because several xenobiotics known to modulate the risk for developing CRC can modulate BT transport, it is plausible that interference with BT transport contributes to their anticarcinogenic or procarcinogenic effect and that these compounds may interfere with the anticarcinogenic effect of BT.
Moreover, differences in MCT1, SMCT1 and BCRP expression between normal colonocytes and tumoral cells contribute to the different effects of BT in these cells (‘the BT paradox’). More specifically, BT is transported into normal colonic epithelial cells by both MCT1 and SMCT1, but its intracellular concentration is kept low because it is efficiently metabolized and effluxed from these cells by BCRP-mediated transport. In contrast, colonic epithelial tumoral cells show a decrease in SMCT1 protein expression, and BT is taken up by these cells through MCT1. In these cells, BT accumulates intracellularly because it is inefficiently metabolized (due to the fact that glucose becomes the primary energy source of these cells) and because there is a reduction in BCRP expression.
Conflict of interestNo conflicts of interest are declared by the authors.
Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Portugal (Plano estratégico UID/BIM/04293/2013).