



Adverse drug reactions (ADRs) are a major public health concern and a leading cause of morbidity and mortality in the world. In the case of antiepileptic drugs (AEDs), ADRs constitute a barrier to successful treatment since they decrease treatment adherence and impact patients’ quality of life of patients. Pharmacogenetics aims to identify genetic polymorphisms associated with drug safety. This article presents a review of genes coding for drug metabolising enzymes and drug transporters, and HLA system genes that have been linked to AED-induced ADRs.
DevelopmentTo date, several genetic variations associated with drug safety have been reported: CYP2C9*2 and *3 alleles, which code for enzymes with decreased activity, have been linked to phenytoin (PHT)-induced neurotoxicity; GSTM1 null alleles with hepatotoxicity induced by carbamazepine (CBZ) and valproic acid (VPA); EPHX1 polymorphisms with teratogenesis; ABCC2 genetic variations with CBZ- and VPA-induced neurological ADRs; and HLA alleles (e.g. HLA-B*15:02, -A*31:01, -B*15:11, -C*08:01) with cutaneous ADRs.
ConclusionsPublished findings show that there are ADRs with a pharmacogenetic basis and a high interethnic variability, which indicates a need for future studies in different populations to gather more useful results for larger number of patients. The search for biomarkers that would allow predicting ADRs to AEDs could improve pharmacotherapy for epilepsy.
Las reacciones adversas a medicamentos (RAM) son un problema de salud pública y una importante causa de morbimortalidad a nivel mundial. En el caso de los fármacos antiepilépticos (FAE), la presencia de RAM puede ser un impedimento para lograr el éxito terapéutico al dificultar la adherencia al tratamiento e impactar la calidad de vida del paciente. La farmacogenética busca la identificación de variantes genéticas asociadas a la seguridad de los fármacos. En este artículo se revisan los genes que codifican para enzimas metabolizadoras y transportadores de fármacos, así como en el sistema HLA asociados a RAM inducidas por FAE.
DesarrolloA la fecha, se ha reportado la asociación de los alelos CYP2C9*2 y *3, que codifican para enzimas de actividad reducida, con efectos neurotóxicos por fenitoína (PHT); alelos nulos de GSTM1 asociados con hepatotoxicidad inducida por carbamazepina (CBZ) y ácido valproico (VPA); polimorfismos genéticos de EPHX1 en la teratogénesis inducida por PHT; variantes genéticas de ABCC2 asociadas con RAM neurológicas por CBZ y VPA, y también diversos alelos de HLA (p. ej., HLA-B*15:02, -A*31:01, -B*15:11, -C*08:01) asociados con RAM de tipo cutáneas.
ConclusionesLos hallazgos publicados muestran que existen RAM con base farmacogenética con una alta variabilidad interétnica, lo que refleja la necesidad de que se realicen estudios en distintas poblaciones para poder obtener resultados que sean de utilidad a un número mayor de pacientes. La búsqueda de biomarcadores que permitan la predicción de RAM a FAE podría mejorar la farmacoterapia en la epilepsia.
Pharmacogenetics emerged as a discipline in the 1950s, as a result of several observations of inherited enzymatic deficiencies causing drug toxicity in a specific group of patients. Its main objective has been the study of genetic variations associated with differences in the individual response to drugs.1,2
Pharmacogenetic studies analyse the association of gene allele variants which code for drug metabolising enzymes (DME) and drug transporters and receptors. They also analyse dosage requirements, effectiveness, and presence of adverse drug reactions (ADR). This review focuses on pharmacogenetic advances in the field of ADRs caused by antiepileptic drugs (AED), due to their impact on treatment adherence and quality of life in epileptic patients.
Adverse drug reactionsAdverse drug reactions are a serious problem for patients and public health systems. Incidence of ADRs is estimated at 6.73% in the USA3 and 6.5% in the United Kingdom,4 whereas in Switzerland they represent 3% of deaths.5 ADR prevalence has increased in patients older than 60 years.6 Patients with ADRs may need to be hospitalised or require longer admission times, which increases treatment costs.7-9
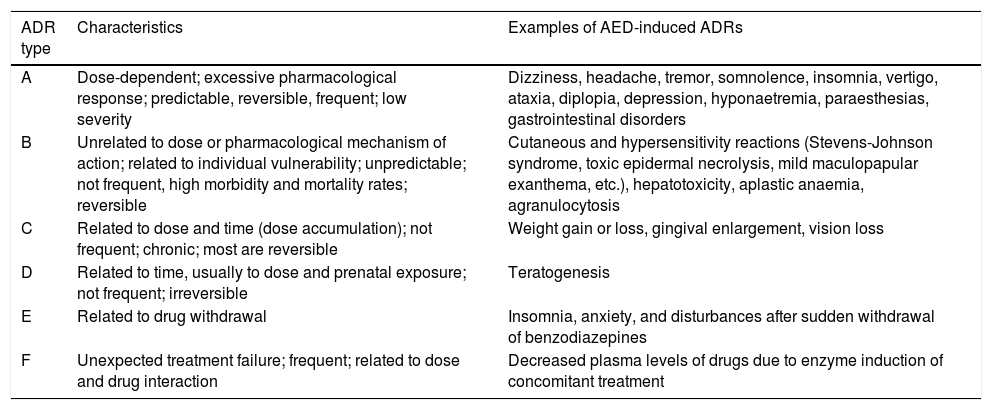
For more than 40 years, the World Health Organization has defined ADRs as “a response to a drug which is noxious and unintended, and which occurs at doses normally used in man for the prophylaxis, diagnosis, or therapy of disease, or for the modifications of physiological function”.10 Researchers have mentioned the need for a new definition to include ADRs caused by medication error, unauthorised use, misuse, and abuse of medicinal products, and the management of ADRs, which includes, for example, the administration of a specific treatment, total discontinuation of the drug, and future precautionary measures, among other considerations.11 Different types of reactions are classified from A to F, with A and B being the most common types. The characteristics of each type are listed in Table 1.11-13
Classification of adverse drug reactions.
| ADR type | Characteristics | Examples of AED-induced ADRs |
|---|---|---|
| A | Dose-dependent; excessive pharmacological response; predictable, reversible, frequent; low severity | Dizziness, headache, tremor, somnolence, insomnia, vertigo, ataxia, diplopia, depression, hyponaetremia, paraesthesias, gastrointestinal disorders |
| B | Unrelated to dose or pharmacological mechanism of action; related to individual vulnerability; unpredictable; not frequent, high morbidity and mortality rates; reversible | Cutaneous and hypersensitivity reactions (Stevens-Johnson syndrome, toxic epidermal necrolysis, mild maculopapular exanthema, etc.), hepatotoxicity, aplastic anaemia, agranulocytosis |
| C | Related to dose and time (dose accumulation); not frequent; chronic; most are reversible | Weight gain or loss, gingival enlargement, vision loss |
| D | Related to time, usually to dose and prenatal exposure; not frequent; irreversible | Teratogenesis |
| E | Related to drug withdrawal | Insomnia, anxiety, and disturbances after sudden withdrawal of benzodiazepines |
| F | Unexpected treatment failure; frequent; related to dose and drug interaction | Decreased plasma levels of drugs due to enzyme induction of concomitant treatment |
Epilepsy is one of the most prevalent neurological disorders globally, affecting approximately 70 million people worldwide and at least 5 million in Latin America.14,15
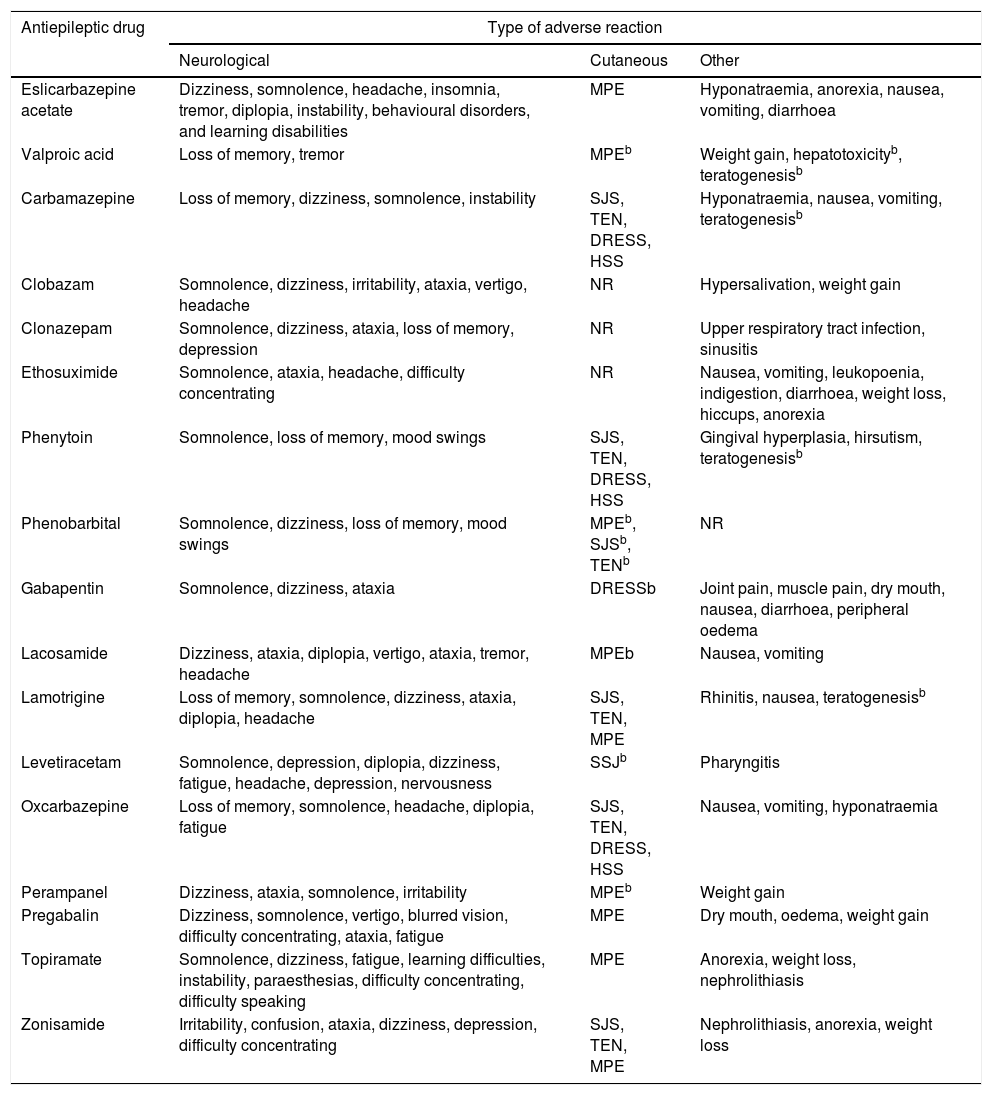
Despite numerous attempts to develop safe, harmless drugs, ADRs are unavoidable. The different mechanisms of action of AEDs may cause undesired effects; these are mainly neurological and psychiatric, although other organs and systems may also be affected (Table 2).16-31
| Antiepileptic drug | Type of adverse reaction | ||
|---|---|---|---|
| Neurological | Cutaneous | Other | |
| Eslicarbazepine acetate | Dizziness, somnolence, headache, insomnia, tremor, diplopia, instability, behavioural disorders, and learning disabilities | MPE | Hyponatraemia, anorexia, nausea, vomiting, diarrhoea |
| Valproic acid | Loss of memory, tremor | MPEb | Weight gain, hepatotoxicityb, teratogenesisb |
| Carbamazepine | Loss of memory, dizziness, somnolence, instability | SJS, TEN, DRESS, HSS | Hyponatraemia, nausea, vomiting, teratogenesisb |
| Clobazam | Somnolence, dizziness, irritability, ataxia, vertigo, headache | NR | Hypersalivation, weight gain |
| Clonazepam | Somnolence, dizziness, ataxia, loss of memory, depression | NR | Upper respiratory tract infection, sinusitis |
| Ethosuximide | Somnolence, ataxia, headache, difficulty concentrating | NR | Nausea, vomiting, leukopoenia, indigestion, diarrhoea, weight loss, hiccups, anorexia |
| Phenytoin | Somnolence, loss of memory, mood swings | SJS, TEN, DRESS, HSS | Gingival hyperplasia, hirsutism, teratogenesisb |
| Phenobarbital | Somnolence, dizziness, loss of memory, mood swings | MPEb, SJSb, TENb | NR |
| Gabapentin | Somnolence, dizziness, ataxia | DRESSb | Joint pain, muscle pain, dry mouth, nausea, diarrhoea, peripheral oedema |
| Lacosamide | Dizziness, ataxia, diplopia, vertigo, ataxia, tremor, headache | MPEb | Nausea, vomiting |
| Lamotrigine | Loss of memory, somnolence, dizziness, ataxia, diplopia, headache | SJS, TEN, MPE | Rhinitis, nausea, teratogenesisb |
| Levetiracetam | Somnolence, depression, diplopia, dizziness, fatigue, headache, depression, nervousness | SSJb | Pharyngitis |
| Oxcarbazepine | Loss of memory, somnolence, headache, diplopia, fatigue | SJS, TEN, DRESS, HSS | Nausea, vomiting, hyponatraemia |
| Perampanel | Dizziness, ataxia, somnolence, irritability | MPEb | Weight gain |
| Pregabalin | Dizziness, somnolence, vertigo, blurred vision, difficulty concentrating, ataxia, fatigue | MPE | Dry mouth, oedema, weight gain |
| Topiramate | Somnolence, dizziness, fatigue, learning difficulties, instability, paraesthesias, difficulty concentrating, difficulty speaking | MPE | Anorexia, weight loss, nephrolithiasis |
| Zonisamide | Irritability, confusion, ataxia, dizziness, depression, difficulty concentrating | SJS, TEN, MPE | Nephrolithiasis, anorexia, weight loss |
DRESS: drug reaction with eosinophilia and systemic symptoms; HSS: hypersensitivity syndrome; MPE: maculopapular exanthema; NR: reactions not frequently reported for these antiepileptic drugs; SJS: Stevens-Johnson syndrome; TEN: toxic epidermal necrolysis.
Frequent reactions were those manifesting in ≥1/100 to <1/10 of the patients, and very frequent reactions, those reported by ≥1/10 patients.
These adverse reactions manifest at a frequency of ≥1/100; however, they were included due to their severity and their significance in pharmacogentics.
Taken from Brogden et al.,16 Greenwood,17 Herranz et al.,18 Hill et al.,19 Jarernsiripornkul et al.,20 Ketter et al.,21 Massot et al.,22 Pellock,23 Posner et al.,24 Tomson and Battino,25 Riverol et al.,26 Zaccara et al.27-31
ADRs during AED treatment complicate seizure control and adherence, and contribute to treatment withdrawal in 25% of patients.32-34 In addition to affecting the patient's quality of life,13,35 there is also an economic burden associated with ADRs.36
Pharmacogenetic research assesses the participation of genetic polymorphisms in response variability and susceptibility to specific types of ADR.2,37 The study of these polymorphisms is focused on genes coding for DMEs, AED transporters, and, more recently, on the genes of the human leukocyte antigen (HLA) system.2,38-40
Genetic polymorphisms in drug-metabolising enzymes associated with adverse reactions to antiepileptic drugsPhase I enzymesType A ADRs due to AEDs include neurological and psychiatric effects, vitamin deficiency, endocrine disorders, and hyponatraemia, among others.41 Since these ADRs are dose-dependent, their presence has been associated with polymorphisms in genes coding for DMEs and drug transporters.
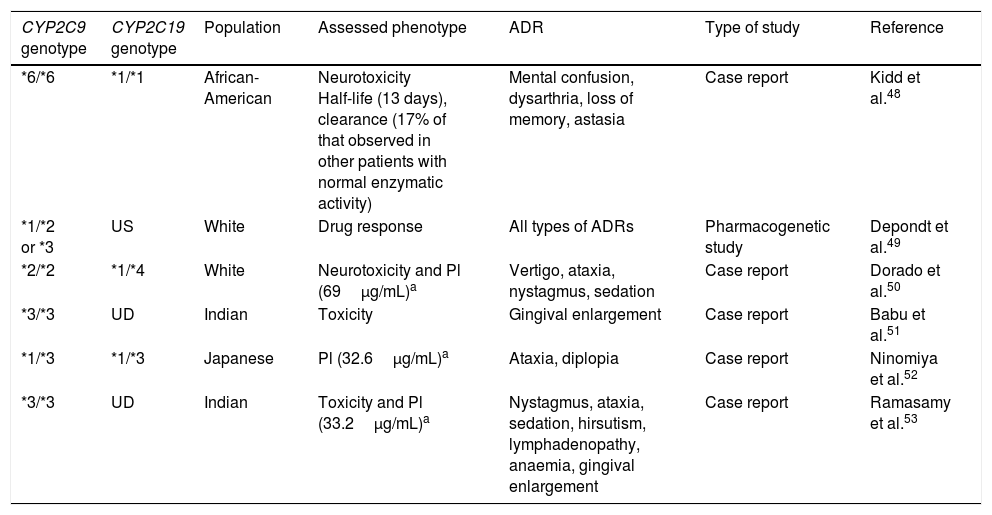
These enzymes are mainly responsible for metabolising endogenous and exogenous compounds. For AEDs, metabolic reactions are catalysed predominantly by cytochrome P450 (CYP) enzymes in phase I metabolism and UDP-glucuronosyltransferase (UGT) enzymes in phase II.42 Two CYP enzymes, CYP2C9 and CYP2C19, are important in the metabolism of phenytoin (PHT), a drug with a narrow therapeutic window and nonlinear pharmacokinetics. CYP2C9 is responsible for 90% of PHT metabolism, while CYP2C19 is responsible for the remaining 10%. Polymorphisms in the genes coding for these cytochromes have been reported to be associated with different ADRs, especially neurological effects. Several allelic variants of CYP2C9 have been described; the 3 most representative and most frequently observed in white subjects are wild-type CYP2C9*1 (Arg144Ile359), CYP2C9*2 (Cys144Ile359), and CYP2C9*3 (Arg144Leu359). The latter 2 variants code for enzymes with decreased activity. Carriers of the CYP2C9*2 genotype present an enzymatic activity of 12% with regards to the wild-type CYP2C9*1, whereas enzymatic activity is only 5% in CYP2C9*3 carriers. In the Spanish population, frequency of CYP2C9*2 and CYP2C9*3 is 16% and 10%, respectively,43 whereas lower frequencies are observed in Chinese and African-American populations, as well as in Mexican mestizo and Mexican indigenous populations.44-46 Studies of African-American populations identified 2 alleles with decreased enzymatic activity: CYP2C9*5 (Ile359Thr) and *6 (c.delA818).47,48 This is important since carriers of these alleles present a poor metaboliser phenotype for PHT, which may cause dizziness, nystagmus, ataxia, excessive sedation, altered level of consciousness, and mental confusion, negatively impacting the patient's quality of life and treatment adherence.49,50 Several reports have described this association (Table 3); however, more studies into different populations, including from Latin America, are necessary to identify the reduced-activity variants specific to each population.
CYP2C9 and CYP2C19 variants associated with the presence of dose-dependent adverse reactions to phenytoin.
| CYP2C9 genotype | CYP2C19 genotype | Population | Assessed phenotype | ADR | Type of study | Reference |
|---|---|---|---|---|---|---|
| *6/*6 | *1/*1 | African-American | Neurotoxicity Half-life (13 days), clearance (17% of that observed in other patients with normal enzymatic activity) | Mental confusion, dysarthria, loss of memory, astasia | Case report | Kidd et al.48 |
| *1/*2 or *3 | US | White | Drug response | All types of ADRs | Pharmacogenetic study | Depondt et al.49 |
| *2/*2 | *1/*4 | White | Neurotoxicity and Pl (69μg/mL)a | Vertigo, ataxia, nystagmus, sedation | Case report | Dorado et al.50 |
| *3/*3 | UD | Indian | Toxicity | Gingival enlargement | Case report | Babu et al.51 |
| *1/*3 | *1/*3 | Japanese | Pl (32.6μg/mL)a | Ataxia, diplopia | Case report | Ninomiya et al.52 |
| *3/*3 | UD | Indian | Toxicity and Pl (33.2μg/mL)a | Nystagmus, ataxia, sedation, hirsutism, lymphadenopathy, anaemia, gingival enlargement | Case report | Ramasamy et al.53 |
Pl: plasma levels; UD: undetermined; US: unspecified; ADR: adverse drug reaction.
Although the Food and Drug Administration (FDA, the regulatory agency in the USA) has not included CYP2C9 polymorphisms in the list of pharmacogenetic biomarkers for PHT response, decreasing the dose prescribed to carriers of one or 2 alleles with decreased enzymatic activity has been considered important, as has monitoring the presence of ADRs and PHT plasma levels.54CYP2C19 also participates in PHT metabolism, although to a lesser extent. CYP2C19 alleles *2A, *2B, *2, *3, *4, *5A, *5B, *6, *7, and *8 present deficient enzymatic activity, potentially increasing the likelihood of toxic effects caused by PHT; however, these have received less study.55,56
Phenytoin metabolism mediated by CYP2C9 and CYP2C19 causes an epoxide type intermediate, which is inactivated by microsomal epoxide hydrolase (EPHX1).57 One study suggested that the teratogenic effects of PHT are due to the formation of this epoxide.58 A study of maternal EPHX1 polymorphisms in women treated with PHT and their children found that the EPHX1 113H and 139R polymorphisms were more frequent in mothers of children with congenital craniofacial abnormalities. Researchers also observed that the maternal EPHX1 Y113/H139 haplotype showed a significant protective effect against craniofacial abnormalities in pregnancies where the mother was receiving PHT.59
Phase II enzymesThe glutathione S-transferase (GST) superfamily of phase II enzymes also participates in AED metabolism. Their participation is important in the biotransformation of carbamazepine (CBZ) into the metabolites responsible for ADRs, such as carbamazepine-10,11-epoxide, arene oxides, and iminoquinones.60 A study in a Japanese population found an association between GSTM1 null alleles and increased levels of alanine aminotransferase and aspartate transaminase in patients treated with CBZ. This finding is associated with CBZ-induced hepatotoxicity, an idiosyncratic and unpredictable ADR which can cause irreversible damage.61 A retrospective study in Japanese patients receiving valproic acid (VPA) reported higher levels of γ-glutamyltransferase in carriers of GSTM1 null alleles.62 Both associations should be confirmed in Japanese and other populations to corroborate the clinical involvement of the GSTM1 null allele in the hepatic damage caused by CBZ and/or VPA.
UGT enzymes play an important role in the removal of potentially toxic lipophilic compounds by forming glucuronides from uridine diphospho-glucuronic acid.63 In the case of AEDs, UGT enzymes play an important role in the metabolism of CBZ, lamotrigine (LTG), oxcarbazepine, and topiramate, among other drugs.64 Some polymorphisms in the genes coding for UGT enzymes have been reported to have an influence on inter-individual variability of the pharmacokinetic parameters of AEDs. For example, differences in LTG plasma levels have been associated with the UGT1A4 L48 V variant65 and differences in VPA plasma levels with the UGT1A3*5 allele.66 A more recent study suggested the association of UGT1A6 552A>C polymorphism with presence of high VPA plasma levels and ADRs induced by this AED, such as ataxia, liver damage, metabolic changes, tremor, hallucinations, pancreatitis, and excessive weight gain.67
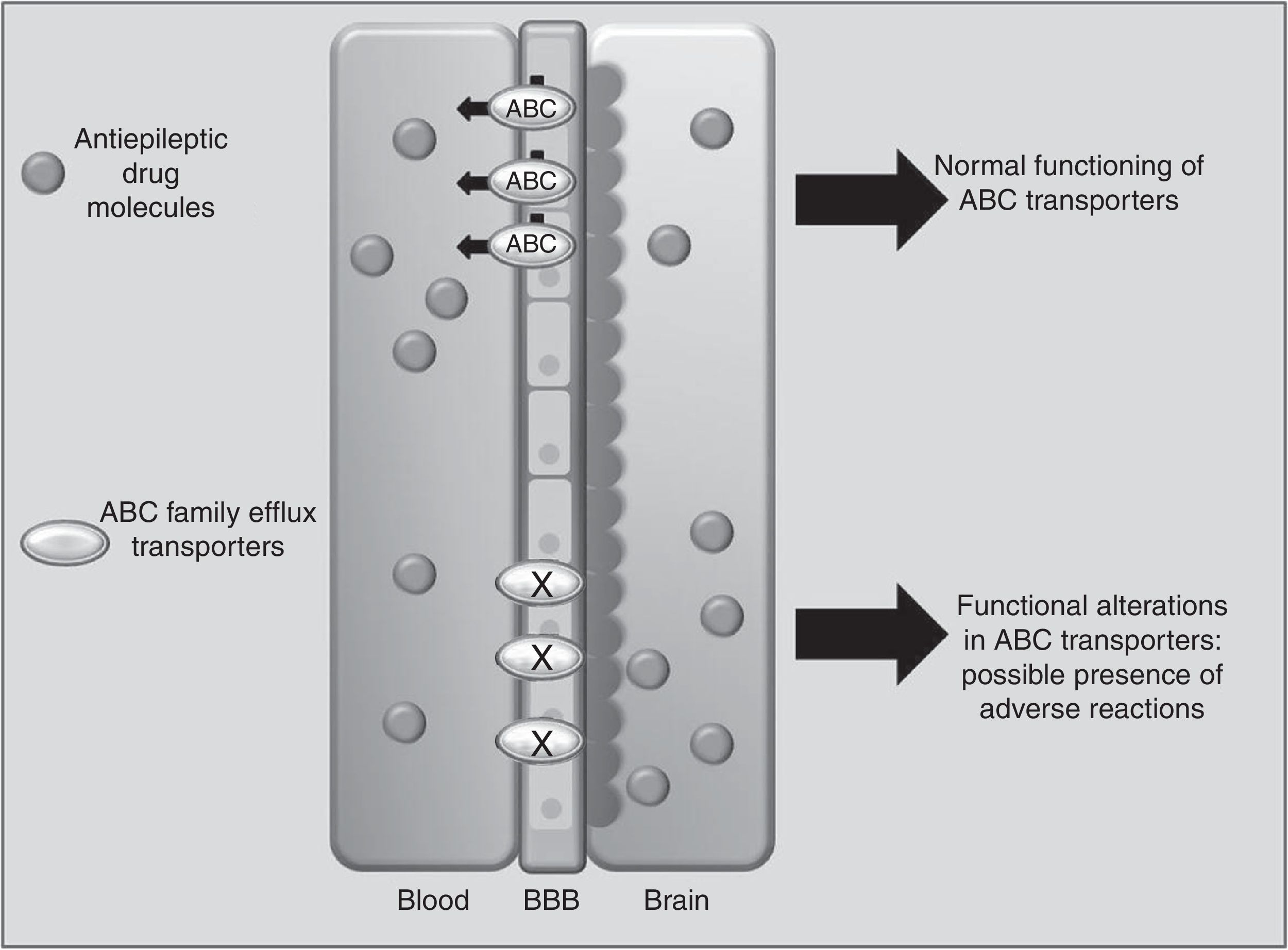
Drug transportersABCC2 is an efflux transporter from the superfamily of ATP-binding cassette (ABC) transmembrane proteins, which utilise the energy released by ATP hydrolysis to translocate solutes and drugs across cellular membranes.68 Such AEDs as PHT, CBZ, and VPA are substrates of this transporter. Researchers have observed that when ABCC2 is overexpressed at the blood-brain barrier, some patients may present refractory temporal lobe epilepsy. In contrast, some polymorphisms of the ABCC2 gene may result in a decrease in the transporter's efflux function, which leads to greater penetration of the drug into the brain, triggering mainly neurological ADRs (Fig. 1).69-71 A study of Korean patients receiving VPA found that the g.-1774delG polymorphism of the ABCC2 gene was associated with presence of ADRs, with patients carrying the G allele being more likely to experience dizziness, headache, somnolence, diplopia, dysarthria, and tremor than those with the deletion allele (P=.0057). Researchers also found that frequency of the T allele at g.-24C>T (rs717620) was higher in the group of patients with neurological ADRs than in the group of patients without these reactions (P=.0274).71 However, other studies in Korean and Japanese populations found no association between ABCC2 polymorphisms and VPA-related ADRs.72,73
 in the development of adverse reactions to antiepileptic drugs.")
Another Korean study of patients receiving CBZ demonstrated an association between the ABCC2 c.1249G>A polymorphism and presence of neurological ADRs. Patients with GA or AA genotypes at this locus reported a higher frequency of neurological ADRs (P=.005).74 An earlier report described that ABCC2 g.-1774delG polymorphism and a haplotype containing the g.-1549G>A, g.-24C>T, c.334-49C>T, and c.3972C>T variations are a predisposing factor for developing liver complications associated with several drugs75; however, these have not been studied in AEDs.
P-glycoprotein (Pgp), coded by the ABCB1 gene, is another transporter of the ABC superfamily, and has been widely studied in the context of AED resistance and pharmacokinetic variability of substrate AEDs of human Pgp, such as PHT, phenobarbital, and LTG.76 Although 3 ABCB1 polymorphisms (3435C>T, 2677G>T/A, and 1236C>T) have been associated with variability of PHT plasma levels,77,78 their association with neurotoxic effects remains unclear.50,79
Cutaneous adverse reactions to antiepileptic drugs and their association with alleles of the HLA systemType B reactions are idiosyncratic, and even though they manifest in a smaller proportion of patients, they result in higher morbidity and mortality rates and require immediate withdrawal of the drug or even an additional treatment to control ADRs.41 These reactions include cutaneous adverse drug reactions (cADR), which have been reported to occur with AEDs and other drug groups. These reactions have an incidence of 10 cases per 1000 new users.80
There are several subtypes of cADRs, with different degrees of severity. Maculopapular exanthema (MPE) is the mildest form, usually involving only the skin and not displaying systemic symptoms; MPE resolves with withdrawal of the drug causing the reaction.81 Drug-induced hypersensitivity syndrome (DIHS) and drug reaction with eosinophilia and systemic symptoms (DRESS) manifest between 3 weeks and 3 months after treatment onset; they are characterised by skin eruption, usually pruritic, by lymphadenopathy, fever of 38-40°C, and reactivation of human herpesvirus 6, which can remain active weeks after the drug is withdrawn.82,83
The most severe forms of cADR are Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), with mortality rates of 5% in SJS and 30% in TEN, and a calculated incidence of 2 cases per million population.84,85 Both syndromes are variants of a single condition, but with different percentages of skin detachment (less than 10% in SJS and more than 30% in TEN).86,87 The most important sequelae affect the eyes: up to 75% of patients with TEN may present such severe ophthalmological complications as blindness.88,89
Although the mechanisms causing cADRs are still to be fully defined, 2 theories have been proposed: the hapten/prohapten theory, and the hypothesis of pharmacological interactions between drugs and immune receptors (p-i). In the first case, the drug molecule, being too small to elicit an immune response, acts as a hapten or prohapten; it forms covalents bonds with endogenous proteins, creating a hapten-carrier complex, and becomes immunogenic.90 The hapten-carrier complex is processed by antigen presenting cells in the major histocompatibility complex (MHC) in the lymph nodes and other tissues, which stimulates T cell production and the subsequent clinical manifestations.91 In contrast, the p-i theory (pharmacological interaction of drugs with immune receptors) suggests that some drugs may bind directly and reversibly (non-covalent bond) to immune receptors, such as the MHC or T cell receptor, stimulating T cells specific to the inducing drug.92
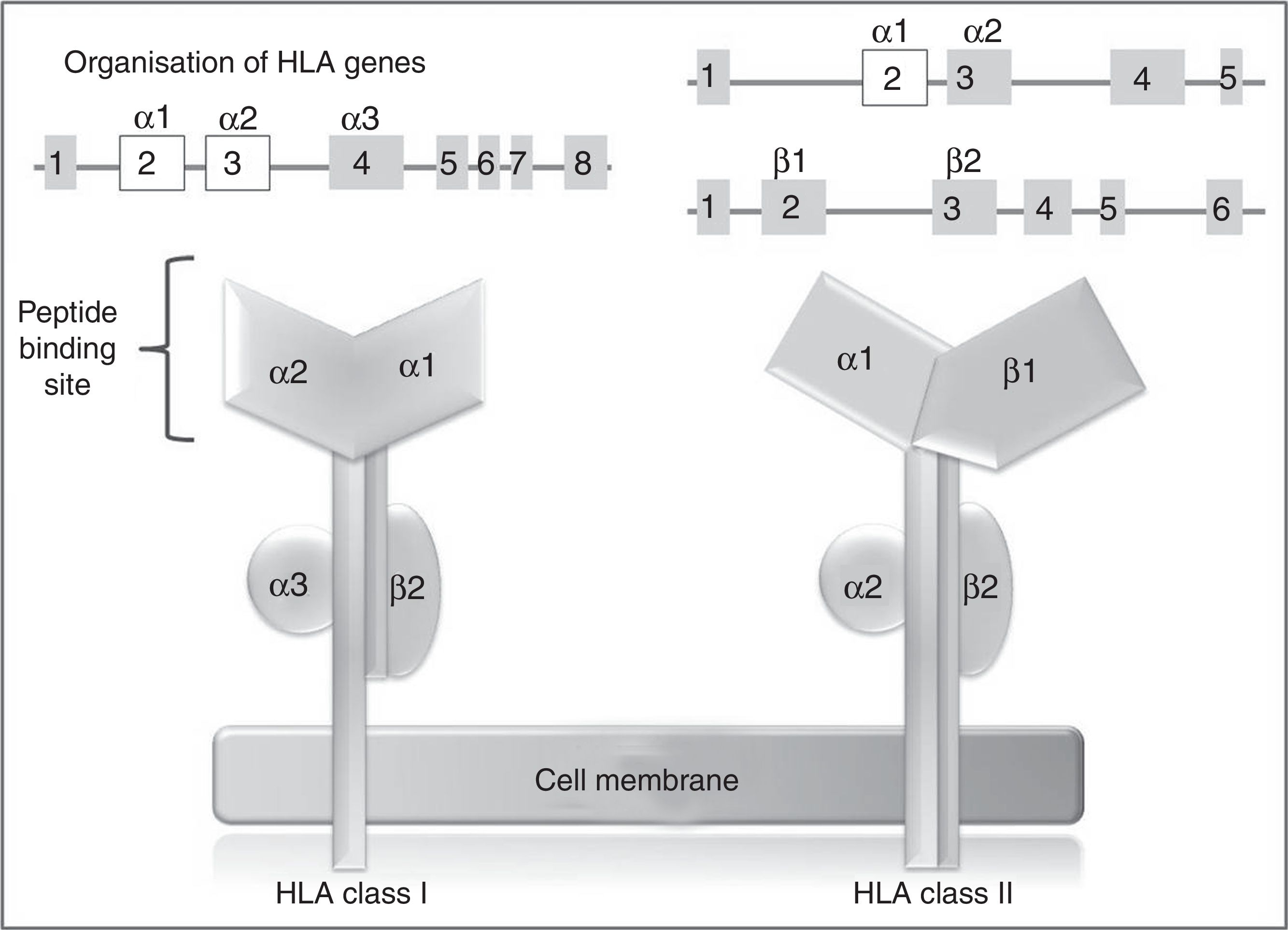
In humans, the genetic region of the MHC is known as the human leukocyte antigen system, and comprises a group of highly polymorphic genes located at 6p21.93 The HLA system is conventionally divided into 3 regions known as classes I, II (classic molecule), and III. The most polymorphic exons in the genes of the classic molecule are those coding for the binding sites for peptides of class I and II HLA molecules94 (Fig. 2). Therefore, pharmacogenetic research has focused in recent years on the identification of class I and (to a lesser extent) class II HLA alleles associated with cADRs (Table 4).
 exons are shown as squares and introns as lines; above the squares are labels indicating the HLA molecule domains (depicted below) coded for by each exon; exons shown in white are those that contain the majority of the polymorphisms included in these genes.")
HLA class I and II genes and alleles: organisation of HLA system genes; (above) exons are shown as squares and introns as lines; above the squares are labels indicating the HLA molecule domains (depicted below) coded for by each exon; exons shown in white are those that contain the majority of the polymorphisms included in these genes.
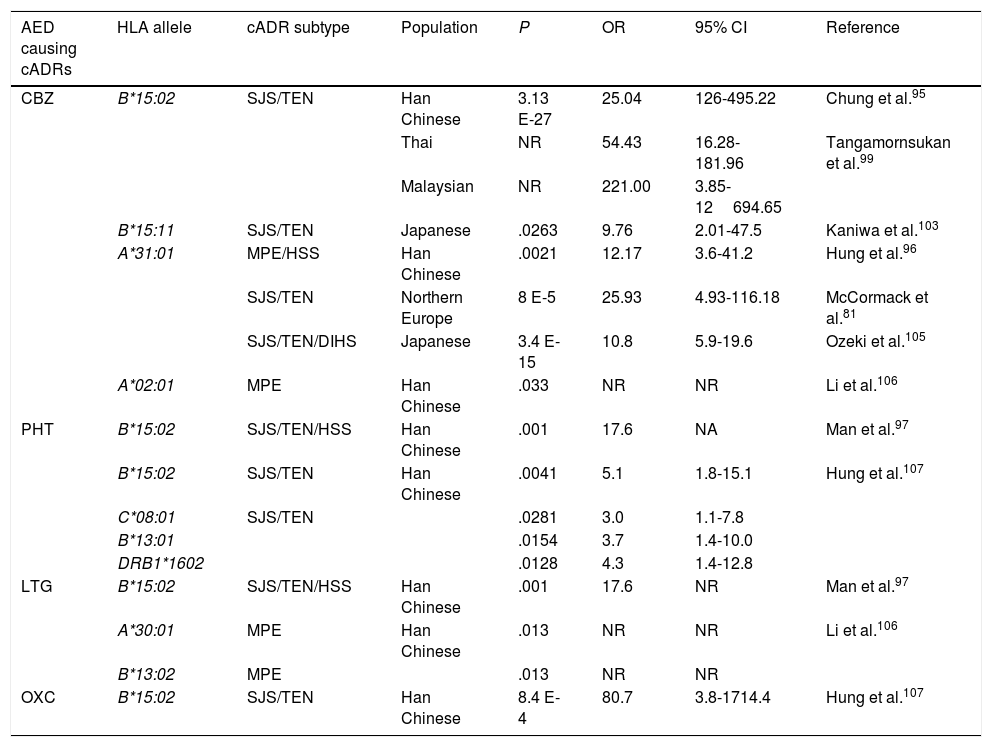
HLA alleles associated with cutaneous adverse reactions caused by antiepileptic drugs.a
| AED causing cADRs | HLA allele | cADR subtype | Population | P | OR | 95% CI | Reference |
|---|---|---|---|---|---|---|---|
| CBZ | B*15:02 | SJS/TEN | Han Chinese | 3.13 E-27 | 25.04 | 126-495.22 | Chung et al.95 |
| Thai | NR | 54.43 | 16.28-181.96 | Tangamornsukan et al.99 | |||
| Malaysian | NR | 221.00 | 3.85-12694.65 | ||||
| B*15:11 | SJS/TEN | Japanese | .0263 | 9.76 | 2.01-47.5 | Kaniwa et al.103 | |
| A*31:01 | MPE/HSS | Han Chinese | .0021 | 12.17 | 3.6-41.2 | Hung et al.96 | |
| SJS/TEN | Northern Europe | 8 E-5 | 25.93 | 4.93-116.18 | McCormack et al.81 | ||
| SJS/TEN/DIHS | Japanese | 3.4 E-15 | 10.8 | 5.9-19.6 | Ozeki et al.105 | ||
| A*02:01 | MPE | Han Chinese | .033 | NR | NR | Li et al.106 | |
| PHT | B*15:02 | SJS/TEN/HSS | Han Chinese | .001 | 17.6 | NA | Man et al.97 |
| B*15:02 | SJS/TEN | Han Chinese | .0041 | 5.1 | 1.8-15.1 | Hung et al.107 | |
| C*08:01 | SJS/TEN | .0281 | 3.0 | 1.1-7.8 | |||
| B*13:01 | .0154 | 3.7 | 1.4-10.0 | ||||
| DRB1*1602 | .0128 | 4.3 | 1.4-12.8 | ||||
| LTG | B*15:02 | SJS/TEN/HSS | Han Chinese | .001 | 17.6 | NR | Man et al.97 |
| A*30:01 | MPE | Han Chinese | .013 | NR | NR | Li et al.106 | |
| B*13:02 | MPE | .013 | NR | NR | |||
| OXC | B*15:02 | SJS/TEN | Han Chinese | 8.4 E-4 | 80.7 | 3.8-1714.4 | Hung et al.107 |
CBZ: carbamazepine; DIHS: drug-induced hypersensitivity syndrome; AED: antiepileptic drug; HLA: human leukocyte antigen; HSS: hypersensitivity syndrome; CI: confidence interval; LTG: lamotrigine; MPE: maculopapular exanthema; NR: not reported; OR: odds ratio; OXC: oxcarbazepine; PHT: phenytoin; cADR: cutaneous adverse drug reaction; SJS: Stevens-Johnson syndrome; TEN: toxic epidermal necrolysis.
In 2004, Chung et al.95 showed a strong association between the HLA-B*15:02 allele and CBZ-induced SJS in Chinese patients from the province of Han. All patients with SJS (n=44) tested positive for this allele, while only 3% of CBZ-tolerant patients (n=101) and 8.6% of the healthy individuals (n=93) carried the HLA-B*15:02 allele (OR: 2.504, 95% CI, 1.26-49.522, P<.001). This association has been confirmed by other studies in patients from Han province, as well as from central and south-east China,96-98 Malaysia, and Thailand.99 This important finding led the FDA to include this information in the labelling of CBZ packaging and to recommend genetic testing for HLA alleles before starting treatment in patients of Asian descent.100 In contrast, other studies have shown that this allele is not universal and depends upon the study population. Thus, no association was found between the HLA-B*15:02 allele and patients with SJS in white and Japanese populations101,102; however, the frequency of the HLA-B*15:11 allele was reported to be higher in Japanese patients with SJS than in the general population.103
An association has also been reported between the HLA-A*31:01 allele and MPE or hypersensitivity syndrome (HSS) induced by CBZ in Chinese patients from Han province; the allele was found in 25.8% of patients with cADRs and only 2.8% of CBZ-tolerant patients.96 This same allele was found to be associated with CBZ-induced cADRs in patients from northern European countries,81 but no association has been found with cADRs induced by PHT or LTG.104
Similarly, 3 HLA haplotypes associated with AED-induced cADRs have been reported: the 8.1 ancestral haplotype HLA-A*01:01/-Cw*07:01/-B*08:01/-DRB1*03:01/-DQA1*05:01/-DQB1*02:01, associated with CBZ-induced HSS in white populations108; and HLA-A*24:01/-B*59:01/-C*01:02 and HLA-A*02:01/-B*15:18/-C*07:04, with high relative risk values for severe cADRs induced by CBZ in Japanese patients (16.09 and 28.94, respectively).109
The association of genetic variants close to the HLA-E locus (rs1264511) and the motilin (rs2894342) and the CYP2B6 genes (rs1042389) with the presence of CBZ-induced cADRs has also been reported in a Han Chinese population; however, these results were not robust.96
ConclusionThe data reviewed show that interethnic variability has a strong effect on the association of genetic polymorphisms with presence of AED-induced ADRs. To date, research into the HLA-B*15:02 allele and its relationship with CBZ-induced cADRs in patients of Asian birth or descent has been essential in the pharmacogenetic study of ADRs induced by antiepileptics. Therefore, such regulatory agencies as the FDA have considered this allele a pharmacogenetic biomarker for these populations. Nevertheless, there continues to be controversy regarding the replication of results in other populations, including Asian populations.
Another important finding in the pharmacogenetics of AEDs is the association of CYP2C9*2 and *3 alleles with the development of neurological ADRs due to decreased enzymatic activity of CYP2C9. Furthermore, ABCC2 variants are gaining importance in the presence of ADRs since they participate in the transport of AEDs across the blood-brain barrier; however, this association has not been demonstrated sufficiently. Assessing the participation of genes coding for AED receptors, such as SCN1A, SCN2A, and GABRA1, in the appearance of ADRs may also be an interesting line of research.
The identification of pharmacogenetic biomarkers enabling undesired effects of AEDs to be predicted would help increase safety when prescribing these drugs.
FundingThis review article was drafted as part of a project funded by the Consejo Nacional de Ciencia y Tecnología (CONACYT #167261) of Mexico and the doctoral research grant (CONACYT #369708) awarded to Ingrid Fricke-Galindo. This study was coordinated within the Red Iberoamericana de Farmacogenética y Farmacogenómica (SIFF-RIBEF www.ribef.com).
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fricke-Galindo I, Jung-Cook H, LLerena A, López-López M. Farmacogenética de reacciones adversas a fármacos antiepilépticos. Neurología. 2018;33:165–176.
