



Sporadic Creutzfeldt-Jakob disease (CJD) is the most frequent form of presentation of prion diseases. Although the clinical characteristics of this condition are well known,1,2 lower motor neuron involvement is a rare form of presentation, especially in the early stages of the disease.3
Our patient was a healthy 54-year-old man with a 4-month history of poor coordination and gait instability. The neurological examination detected fasciculations in the deltoid, biceps, triceps, and quadriceps muscles, associated with amyotrophy and mild weakness of the proximal muscles of his limbs, especially the quadriceps, deltoid, and periscapular muscles. The patient also displayed global areflexia with mild dysmetria in all limbs, predominantly affecting the left side; orthostatic tremor; and ataxic gait with inability to walk in tandem. He showed no signs of upper motor neuron involvement.
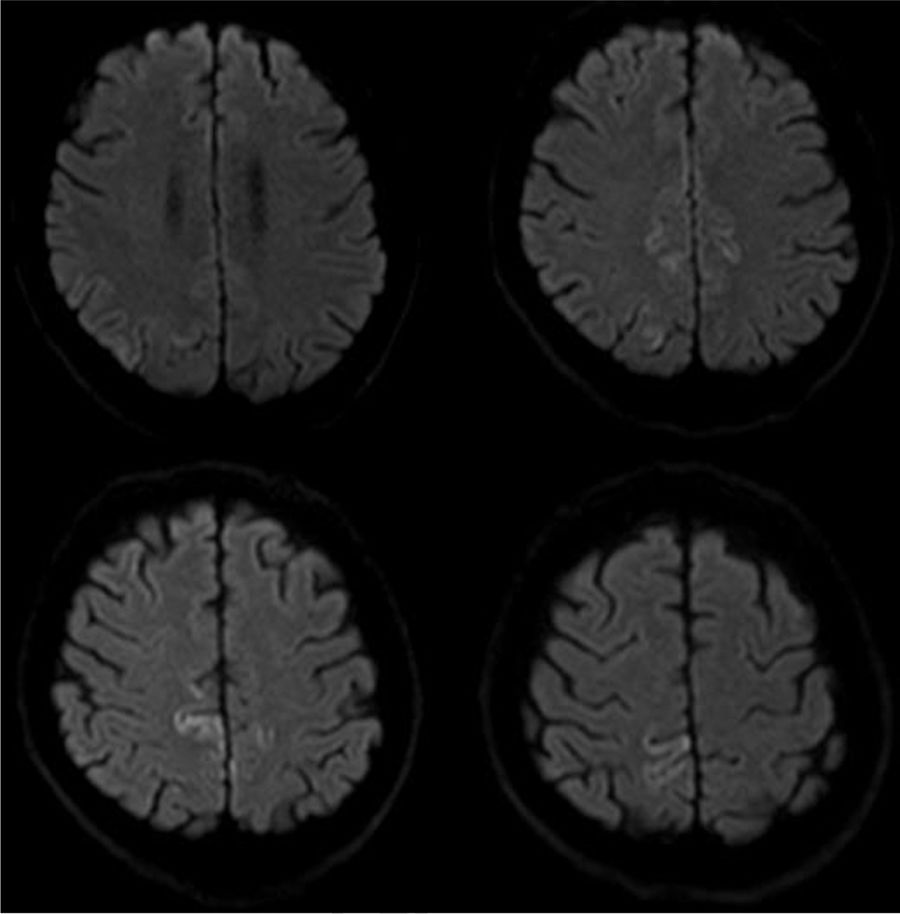
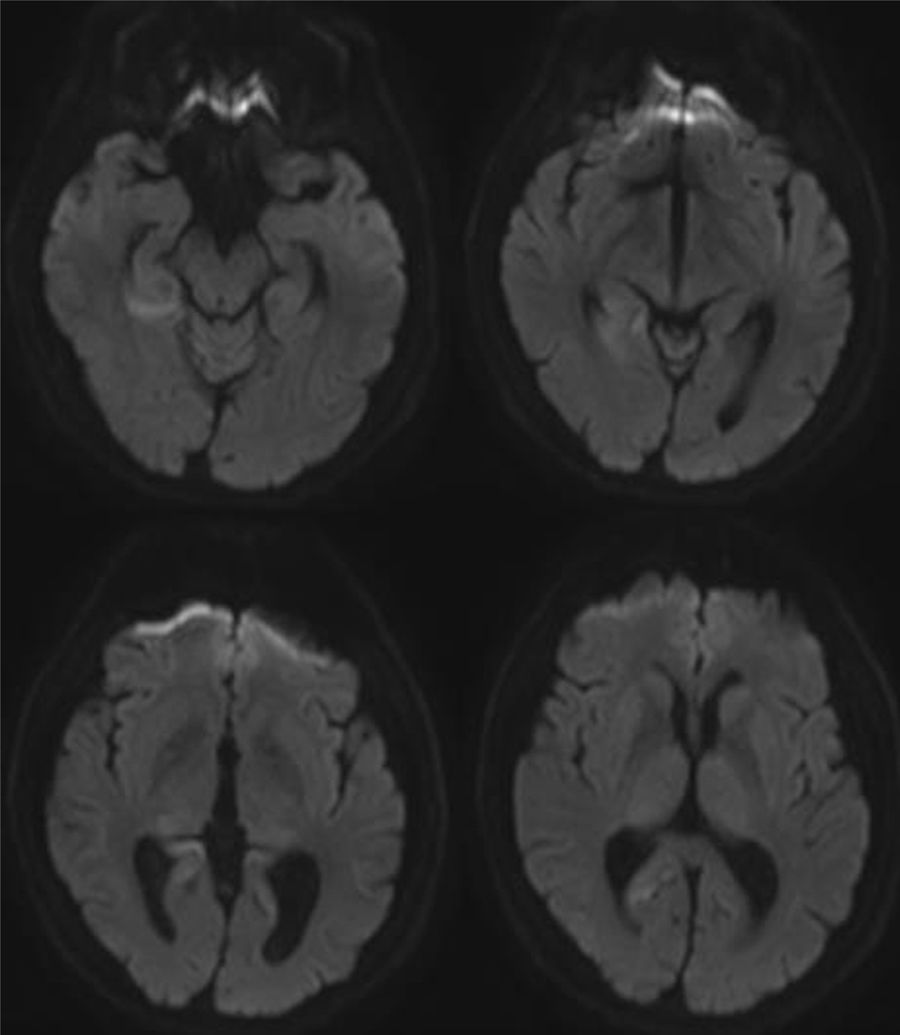
A brain MRI scan revealed cortical alterations on diffusion-weighted sequences only (Fig. 1). Laboratory tests included a complete blood count; a biochemical study; kidney, liver, and thyroid function tests; and vitamin B12 and B9 determination. Results were normal. The tests for infection, paraneoplastic antibodies, tumour markers, and markers of autoimmunity yielded negative results. An electroencephalography revealed mild, diffuse disorganisation of background activity; electromyography showed extensive denervation in the form of fibrillations, positive waves, and fasciculations in all muscles explored. The nerve conduction study revealed no abnormalities. A neuropsychological study revealed mild visuospatial alterations, short-term memory deficits, and reduced verbal fluency; these findings are compatible with cortico-subcortical cognitive impairment. CSF analysis yielded positive results for 14-3-3 protein and elevated tau protein levels. A brain MRI scan performed one month later revealed additional signal alterations in the cortex in diffusion-weighted sequences (Fig. 2). The patient's clinical status progressed to severe cognitive impairment and disabling ataxia; fasciculations were visible in distal muscles of the limbs, with no involvement of face or tongue muscles. In the final stages, he developed akinetic mutism, and died 15 months after diagnosis.
 and in the right superior parietal lobule at the parasagittal level.")

Sporadic CJD is characterised by rapidly progressing dementia, ataxia, muscle tone alterations, and myoclonus.3 However, patients may not develop all these symptoms, and they may present atypically. Our patient displayed amyotrophy, fasciculations, and arreflexia4,5; presentation of these symptoms at disease onset is extremely rare.6,7 The pathophysiological mechanisms of this rare form of presentation are difficult to determine given that post mortem studies only examine the brain; neural loss in the anterior horn of the spinal cord secondary to spongiform degeneration has been proposed as the causal mechanism.4,7,8 Amyotrophic CJD was once regarded as a specific form of the disease.9 Lower motor neuron involvement has been described in familial and sporadic forms of the disease,6 but has not been associated with a specific genetic alteration; the only case in the literature undergoing genotyping had the 129 Met/Met variant.6
Imaging studies reveal isolated cortical hyperintensity in up to one-third of patients with a final diagnosis of sporadic CJD.10 As in our case, the available evidence suggests that diffusion-weighted sequences are the most sensitive for detecting spongiform changes in the brain secondary to CJD, particularly during the early stages of the disease.11–13
Although motor neuron involvement led us to broaden the differential diagnosis, neuroimaging findings and the presence of cerebellar involvement and mild cognitive impairment enabled early aetiological diagnosis. Copresence of amyotrophic lateral sclerosis is unlikely in view of the absence of signs of upper motor neuron involvement, the lack of bulbar symptoms, and the rapid progression of symptoms.
In conclusion, although lower motor neuron involvement is rare in patients with sporadic CJD, it should be included within the clinical spectrum of the condition; lower motor neuron involvement as an early manifestation of the disease may enable early diagnosis.
FundingNo funding was received for this study.
Please cite this article as: Díaz-Díaz A, Hervás-García M, Amela-Peris R, García-Rodríguez JR. Amiotrofia y fasciculaciones como forma de presentación de la enfermedad de Creutzfeldt-Jakob esporádica. A propósito de un caso. Neurología. 2019;34:65–67.
