




Alzheimer disease (AD) is the main cortical neurodegenerative disease. The incidence of this disease increases with age, causing significant medical, social and economic problems, especially in countries with ageing populations.
ObjectiveThis review aims to highlight existing evidence of how vascular dysfunction may contribute to cognitive impairment in AD, as well as the therapeutic possibilities that might arise from this evidence.
DevelopmentThe vascular hypothesis emerged as an alternative to the amyloid cascade hypothesis as an explanation for the pathophysiology of AD. This hypothesis locates blood vessels as the origin for a variety of pathogenic pathways that lead to neuronal damage and dementia. Destruction of the organisation of the blood–brain barrier, decreased cerebral blood flow, and the establishment of an inflammatory context would thus be responsible for any subsequent neuronal damage since these factors promote aggregation of β-amyloid peptide in the brain. The link between neurodegeneration and vascular dysfunction pathways has provided new drug targets and therapeutic approaches that will add to the treatments for AD.
ConclusionsIt is difficult to determine whether the vascular component in AD is the cause or the effect of the disease, but there is no doubt that vascular pathology has an important relationship with AD. Vascular dysfunction is likely to act synergistically with neurodegenerative changes in a cycle that exacerbates the cognitive impairment found in AD.
La enfermedad de Alzheimer (EA) es la principal enfermedad neurodegenerativa cortical. Su incidencia aumenta con la edad, lo que provoca importantes problemas médicos, sociales y económicos, especialmente en países con población envejecida.
ObjetivoEl objetivo de esta revisión es poner de manifiesto las evidencias que existen sobre el modo en que la disfunción vascular puede contribuir al deterioro cognitivo en la EA, así como las posibilidades terapéuticas que de ello podrían derivarse.
DesarrolloLa hipótesis vascular ha surgido como alternativa a la hipótesis de la cascada amiloide como explicación de la fisiopatología de la EA. Esta hipótesis sitúa en los vasos sanguíneos el origen de una serie de estímulos patogénicos que llevarían a la lesión neuronal y la demencia. La destrucción de la organización de la barrera hematoencefálica, la disminución del flujo sanguíneo cerebral y el establecimiento de un contexto inflamatorio serían responsables de un consecuente daño neuronal a causa de favorecer la agregación del péptido β-amiloide en el cerebro. Las vías que relacionan la disfunción vascular con la neurodegeneración han proporcionado nuevos enfoques terapéuticos y dianas farmacológicas con las que avanzar en la búsqueda de tratamientos para la EA.
ConclusionesResulta difícil determinar si el componente vascular de la EA es la causa o el efecto de la enfermedad, pero no cabe duda de que la enfermedad vascular tiene una relación importante con la EA. Es probable que la disfunción vascular actúe sinérgicamente con los cambios neurodegenerativos en un ciclo que agrava el deterioro cognitivo propio de la EA.
Alzheimer disease (AD) is the main neurodegenerative disease affecting the cortex; its principal clinical manifestation is dementia, or progressive loss of cognitive function.1 Cognitive impairment in AD usually follows a characteristic clinical course, starting with memory deficits which usually go unnoticed but which soon begin to affect daily life activities. In intermediate stages of AD, patients are no longer able to work, get lost easily, feel confused, and need constant supervision. Language impairment appears: it first affects naming, followed by comprehension and verbal fluency. These patients also have apraxia and difficulty performing numerous motor tasks. Wandering, impaired reasoning, and delirium appear in late stages of the disease. Patients with terminal AD are stiff, mute, and unable to carry out physiological functions without assistance.2
According to a study by Ferri et al.,3 over 23.4 million people have dementia today and 4.6 new cases are diagnosed each year, which is equivalent to a new case every 7seconds. It is believed that these figures will double every 20 years; by 2040, there will be 81.1 million people with dementia. Patients rarely develop symptoms before the age of 50, but the incidence of the disease increases with age. This progressive increase has led to medical, social, and economic problems, especially in countries with accentuated population ageing.1 As for Spain, the prevalence of AD is estimated at 7.7% in people older than 70; the disease is slightly more frequent in women than in men.4 In 2013, AD was the seventh most common cause of death and the most common type of dementia in the Spanish population.5
In view of the high prevalence of AD and its considerable medical, sociological, and economic impact, we aim to review available evidence on the mechanism by which vascular disease contributes to cognitive impairment in AD and cover the potential treatment options.
The amyloid cascade hypothesis in Alzheimer diseaseIn 1907, psychiatrist Alois Alzheimer first described the pathognomonic signs of AD in Auguste D., a 51-year-old woman who had been admitted to a mental institution due to symptoms of dementia. Histopathology studies of her brain revealed plaques and neurofibrillary tangles; Alzheimer stated that dementia symptoms in his patient were linked to these lesions.6,7
Many years later, in 1984, the amyloid cascade hypothesis was proposed after Glenner and Wong8 found the amyloid-β peptide (Aβ) to be the main component of the plaques that Alzheimer had observed. Three years later, in 1987, Goldgaber et al.9 located the gene coding for the amyloid precursor protein (APP), which generates Aβ, on chromosome 21. It was later determined that AD may be transmitted by autosomal dominant inheritance of a mutation in the gene coding for APP.10 Together, these observations formed the amyloid cascade hypothesis which synthesises currently available histopathological and genetic data on AD. The hypothesis was fully developed by Hardy and Higgings11 in 1992 and has been revised and updated on several occasions since then.12
During the 1990s, cumulative experimental evidence supported the hypothesis that APP and the proteolytic processing of this protein played a major role in the pathophysiology of AD. Researchers have described several mutations related to the disease in sites within APP that are normally cleaved by a series of proteases that were identified as α-, β-, and γ-secretases.13 Another turning point in amyloid research was the finding that Aβ generation depended on APP proteolytic cleavage by β- and γ-secretases.14,15 On the other hand, several researchers observed in 1995 that AD may also be caused by autosomal dominant mutations of the presenilin-1 (PTEN-1) and presenilin-2 (PTEN-2) genes located on chromosomes 14 and 1, respectively.16,17 However, it was Wolfe et al.18 who in 1999 demonstrated that these 2 homologous proteins constitute the catalytic site of γ-secretase and are therefore essential to its proteolytic function. This finding confirmed the effect of secretases on APP and their fundamental role in AD pathophysiology.
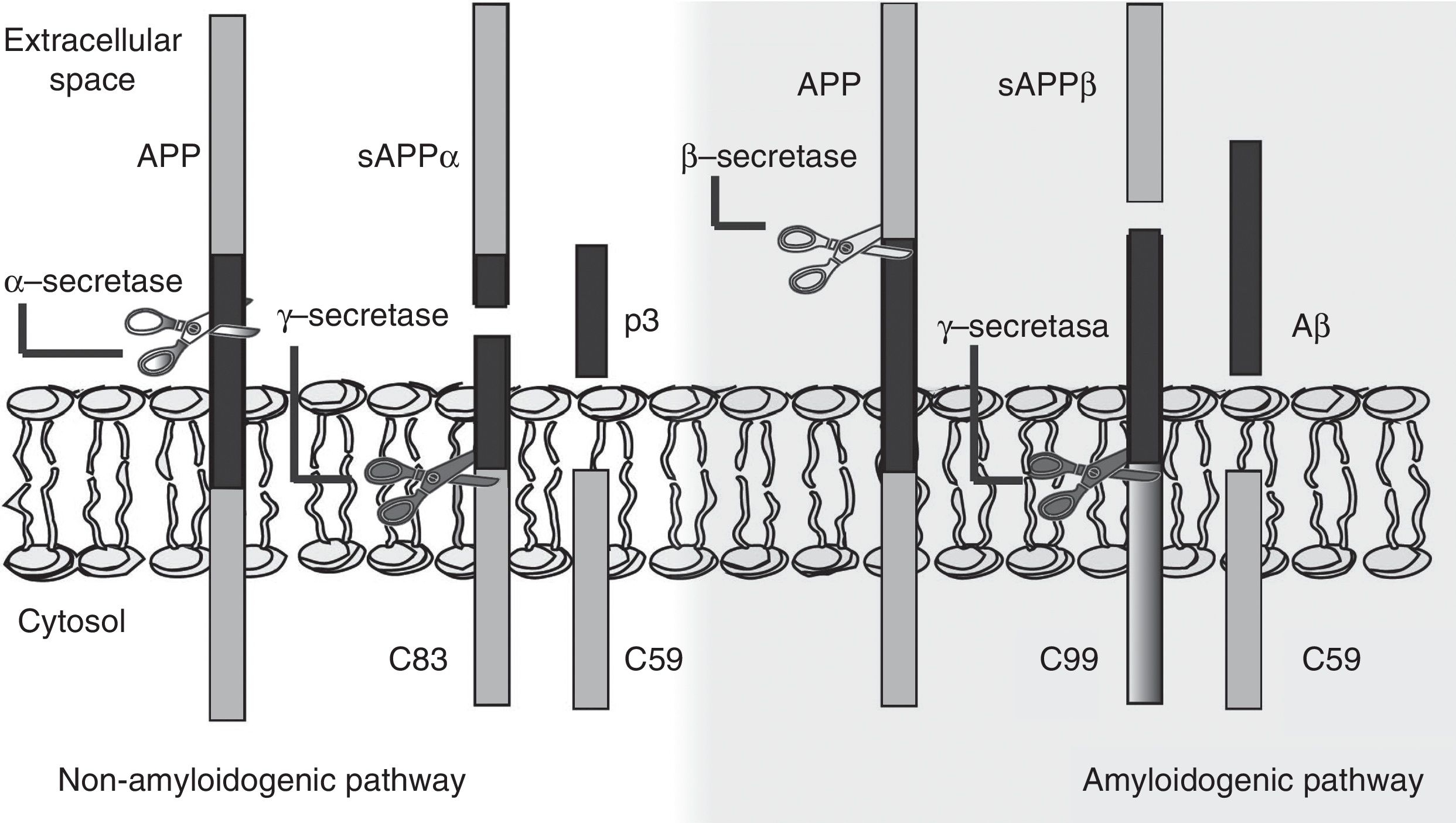
The amyloid cascade hypothesis posits that Aβ amyloid production is responsible for neuronal dysfunction and death, leading to neurodegeneration and dementia.19 It is therefore essential to determine the mechanism leading to Aβ degeneration from APP, a single-pass transmembrane protein with a large extracellular domain and containing 770 amino acids. It is widely expressed in neurons but its physiological function has yet to be fully understood.20 The amyloid cascade hypothesis proposes that APP may be processed by either the non-amyloidogenic or the amyloidogenic pathway (Fig. 1). The most frequent type of APP processing follows the non-amyloidogenic pathway and involves α-secretase, which cleaves APP 83 amino acids from the C-terminus, resulting in an N-terminal fragment that is secreted into the extracellular domain (sAPPα) and a C-terminal fragment of 83 amino acids (C83); the latter remains associated with the membrane and subsequently cleaved by γ-secretase, yielding a short soluble peptide (p3). In the amyloidogenic pathway, however, APP is first cleaved by β-secretase at the position 99 aminoacids from the C-terminus. This generates a secreted extracellular domain (sAPPβ) and a membrane-associated C-terminal fragment containing 99 amino acids (C99). C99 undergoes further proteolytic cleavage by γ-secretase at different locations, releasing Aβ, which contains between 37 and 49 amino acids (Aβ37-Aβ49); Aβ40 is the most frequently released fragment. These Aβ peptides can aggregate into small neurotoxic oligomeric structures, eventually forming the senile plaques typically seen in AD.21,22
 processing: non-amyloidogenic pathway and amyloidogenic pathway. The amyloidogenic pathway promotes amyloid-β peptide (Aβ) generation and senile plaque formation.")
Today, the apolipoprotein E gene (APOE), and more specifically the APOE ¿4 allele, is known to be the main genetic risk factor for AD.23 Humans have 3 common APOE alleles: APOE ¿2, APOE ¿3, and APOE ¿4. The role of APOE in AD is not precisely known. It has been hypothesised that these proteins are involved in Aβ degradation; APOE ¿2, APOE ¿3, and APOE ¿4, in that order, are increasingly less effective at clearing Aβ.24 Tiraboschi et al.25 demonstrated that the brains of patients carrying 2 APOE ¿4 alleles showed more marked senile plaque deposition whereas brain tissue with the APOE ¿2 allele had significantly fewer plaques.
In addition to senile plaques, Alois Alzheimer reported the presence of neurofibrillary tangles as a histopathological sign of AD.6 Hyperphosphorylated tau protein is the main component of these structures,26 and this finding has generated growing interest in the role of this protein in AD. Hyperphosphorylated tau protein is a microtubule-associated protein present in the microtubule network of neuronal axons. AD progression leads to tau protein hyperphosphorylation, which impairs that protein's ability to bind to and stabilise microtubules. This results in decreased axonal transport and subsequent neuronal dysfunction and death. There may be a connection between Aβ and tau protein, although it has not been clearly identified.27
The vascular hypothesis of Alzheimer diseaseThe vascular hypothesis of AD is an alternative to the amyloid cascade hypothesis for explaining the pathogenesis of AD. It was postulated in 1993 by de la Torre and Mussivand.28 These authors observed a reduction, which was proportional to disease severity, in cerebral blood flow, glucose metabolism, and oxygen consumption in patients with AD.
Today, the vascular hypothesis is supported by increasing evidence that vascular dysfunction plays a major role in AD development. Senile plaque and neurofibrillary tangles may be the result, rather than the cause, of neurodegeneration. These structures have been observed in histology studies of the brains of patients experiencing traumatic brain injury, which suggests that APP overexpression and amyloid deposition may occur during the phase of acute response to neuronal injury.29 Moreover, the degree of Aβ deposition does not seem be correlated with the level of severity of cognitive dysfunction in patients with AD. In fact, amyloid deposition has also been found in the brains of cognitively healthy individuals.30
Many studies support the association between different vascular risk factors and an increased likelihood of developing AD. Launer et al.31 concluded that arterial hypertension increased the risk of AD in elderly men who had never received antihypertensive drugs. Kivipelto et al.32 demonstrated that systolic hypertension and high serum cholesterol levels in middle-aged people increased the risk of developing AD later in life. Obesity has also been linked to increased risk of AD. The results of the study by Xu et al.33 support the hypothesis that overweight and obesity in middle age were independently associated with increased risk of AD. Insulin resistance is one of the main obesity-linked risk factors for AD.34,35 In fact, it plays a major role in the pathophysiology of type 2 diabetes mellitus, found by multiple epidemiological studies to increase the risk of AD.36,37 Atherosclerosis is associated with AD according to several studies including that by Hofman et al.38; these authors found that patients with atherosclerosis were 3 times more likely to develop AD. Another recent postulate is that both vascular risk factors and genetic risk factors may contribute to AD. Rodrigue et al.39 hypothesised that patients with a vascular risk factor (hypertension) plus a genetic risk factor for AD (presence of APOE ¿4 allele) display more amyloid deposition than individuals without those risk factors. In this study, patients with hypertension and at least one APOE ¿4 allele showed significantly more Aβ deposition compared to those individuals with only one or neither of the risk factors.
Damage to the neurovascular unit in Alzheimer diseaseEndothelial cells, pericytes, neurons, and glial cells form a functional unit called the neurovascular unit. The proximity of different types of non-neuronal cells to one another and to neurons permits effective paracrine regulation, an essential process for normal CNS functioning. Vascular cells, that is, endothelial cells and pericytes, may directly affect neuronal and synaptic function by altering blood flow, permeability of the blood–brain barrier (BBB), nutrient supply, effective degradation of toxic molecules, enzymatic functions, secretion of trophic factors and matrix molecules, expression of vascular receptors, and induction of ectoenzymes.40
Given the BBB's major role in regulating the passage of circulating metabolites to brain tissue, BBB disruption may have a significant impact on the accumulation of neurotoxic molecules in the brain. The levels of many tight junction and adherens junction proteins and/or their adaptor molecules have been found to decrease in AD, leading to alterations in BBB permeability. Expression of matrix metalloproteinase (MMP) also appears to be elevated in patients with neurodegenerative disorders. Tight junction proteins and some extracellular matrix proteins are substrates for MMP; degradation of these proteins may explain the alterations on BBB permeability seen in AD.41 Disruption of BBB permeability may also be explained by alterations in selective transport mechanisms; selective transport is mediated by transporter molecules, which facilitate the transport of certain circulating nutrients to the brain parenchyma through the bloodstream. In line with this idea, a study found low GLUT-1 concentrations in the brain tissue of patients with AD; decreased GLUT-1 expression may reduce glucose transport from blood to the brain parenchyma, which would explain the decrease in the metabolic substrates necessary for neuronal function.42
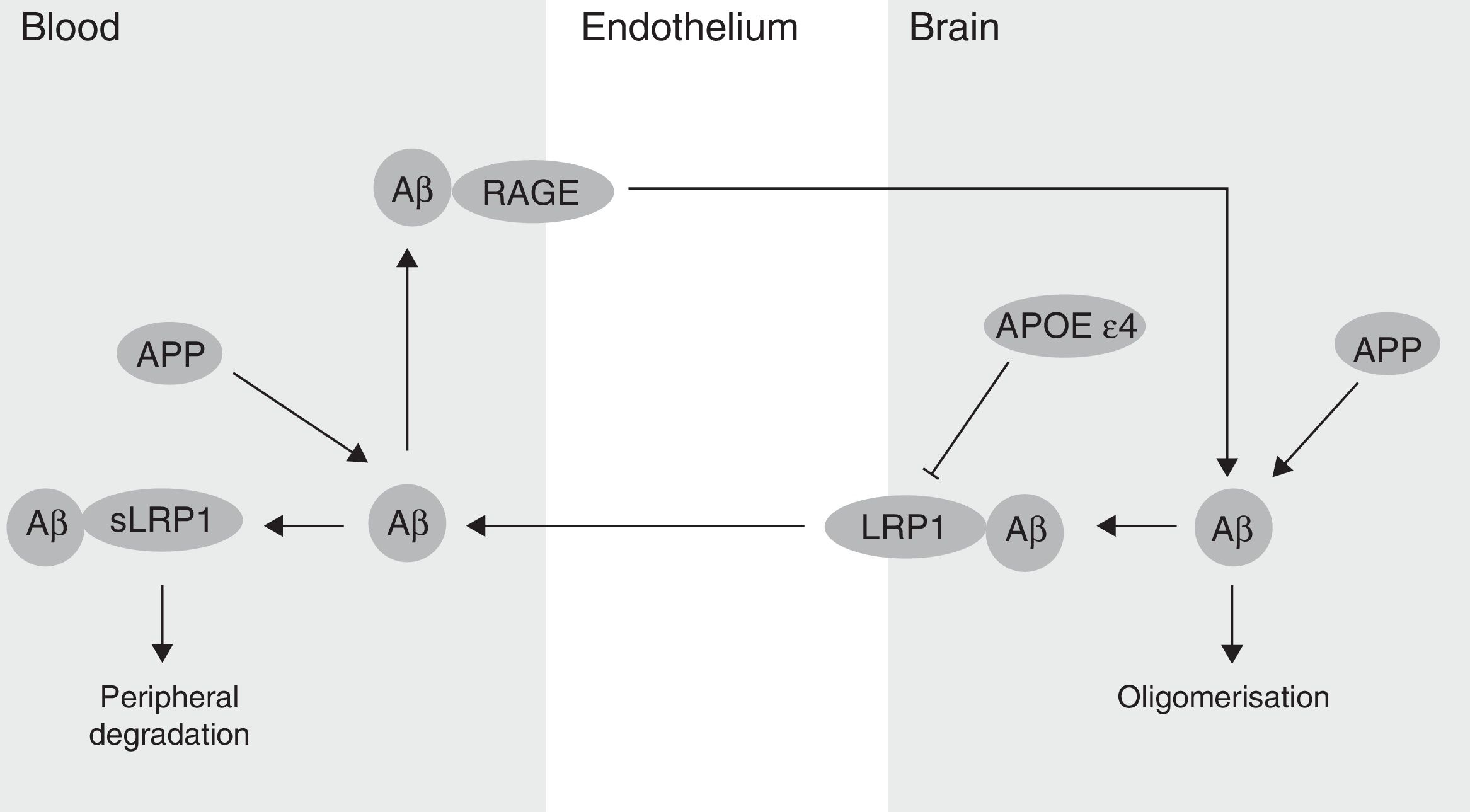
The BBB has been shown to participate in the regulation of Aβ levels in the CNS (Fig. 2). In fact, peripheral Aβ is an important precursor of brain Aβ. Thus, the receptor for advanced glycation end-products (RAGE) regulates Aβ transport to the brain and the propagation of toxicity. RAGE expression in brain endothelium provides a mechanism for influx transport of Aβ and monocytes carrying Aβ across the BBB.41 Studies with animal models of AD have shown that RAGE expression increases in the disease, thereby amplifying the pathogenic responses mediated by Aβ.43 On the other hand, several studies in animal models of AD and in patients have shown decreased Aβ degradation in brain tissue damaged by the disease.44,45 Cell surface receptor LRP1 plays an important role in Aβ degradation.46 Low LRP1 levels in brain microvessels are associated with Aβ accumulation in the brain during ageing and AD.47,48 Brain LRP1 has also been shown to be oxidised in patients with AD by means of an oxidative mechanism that may involve Aβ itself. This process leads to Aβ deposition given that the oxidative form of LRP1 cannot efflux Aβ.49 Lastly, APOE ¿4, unlike APOE ¿3 and APOE ¿2, has been shown to block LRP1-mediated transport of Aβ from the brain, meaning that it promotes Aβ accumulation (Fig. 2).50 Evidence suggests an association between APOE, LRP1, and cholesterol metabolism. Suppressing the expression of LRP1 in forebrain neurons of adult mice has been found to increase brain APOE and cholesterol levels significantly.51
Cerebral blood flow and Alzheimer disease
Experimental evidence suggests that cerebral hypoperfusion precedes the hypometabolism and neurodegeneration that characterise AD.52,53 Chronic cerebral hypoperfusion may result from a number of vascular risk factors, including hypertension, diabetes, atherosclerosis, and heart disease (Fig. 3). These risk factors may affect cerebral vasculature, leading to a progressive decrease in blood supply to the brain.53,54 Vascular tissue dysfunctions found in patients with AD include decreased microvascular density, blood vessel fragmentation and atrophy, increased capillary irregularity, changes in blood vessel diameter, increased thickness of basement membrane, and collagen accumulation in the basement membrane.55 All these changes may cause such neurodegenerative lesions as senile plaques or neurofibrillary tangles as a result of inadequate blood flow to the brain.53,54

Hypoxia, which results from cerebral hypoperfusion, leads to increased expression of hypoxia-inducible factor 1α (HIF-1α) in neurons. HIF-1α binds to the hypoxia-sensitive element of the gene coding for β-secretase, increasing the expression of β-secretase mARN and production of Aβ fragments.56 Likewise, chronic hypoxia has been found to decrease α-secretase expression in human neuroblastoma cells.57 In addition, hypoperfusion may contribute to oxygen deficiencies that can lead to neuroglial metabolic stress; this will increase Aβ production and promote the generation of mitochondrial reactive oxygen species (ROS).58 All these results may help explain the molecular mechanisms underlying the association between chronic cerebral hypoperfusion and accelerated Aβ deposition.59,60
Aβ may also affect cerebral blood flow. Deane et al.61 showed that Aβ interacts with the RAGE receptor on the blood vessel wall. This interaction introduces Aβ into the brain parenchyma and promotes the production of endothelin-1, a vasoconstrictor peptide derived from the endothelium.
Vascular oxidative stress has also been recognised as a key pathogenic factor in AD; it is linked to endothelial dysfunction and decreased brain blood flow (Fig. 3). Oxidative stress leads to endothelial dysfunction as a result of the interaction between superoxide and nitric oxide (NO) in the endothelium to produce peroxynitrite, a potentially harmful oxidant that may cause oxidative damage to proteins, lipids, and nucleic acids. Loss of NO bioavailability as a result of this interaction will decrease NO-mediated vasodilation. Tetrahydrobiopterin, a crucial cofactor for NO synthases, is a target for peroxynitrite-nitrite oxidation; in absence of tetrahydrobiopterin, these enzymes become uncoupled, producing ROS rather than NO which causes further oxidative stress and hinders vasodilation.58 The role of NO in the pathophysiology of AD is gaining increasing interest. Austin et al.62 demonstrated that inhibition, caused by NG-nitro-l-arginine methyl ester (l-NAME), of endothelial nitric oxide synthase (eNOS, an enzyme responsible for endothelial NO synthesis from l-arginine) led to an increase in the levels of APP and β-secretase expression in endothelial cells of human brain microvessels. Likewise, brain tissue of eNOS-deficient mice displayed higher levels of APP and β-secretase and greater β-secretase activity than that of wild-type control mice. These data suggest that endothelial NO plays an important role in the modulation of APP expression and proteolytic processing in brain tissue and microvessels.
In summary, cerebral hypoperfusion promotes Aβ accumulation in the brain parenchyma, and Aβ accumulation in turn aggravates vascular disease. Vascular disease may act in synergy with changes in Aβ levels through a positive feedback system in which tissue damage caused by vascular factors may increase neurodegenerative damage, and vice versa.
Inflammation, vascular dysfunction, and Alzheimer diseaseAlterations in cerebrovascular metabolic functions may also lead to secretion of multiple inflammatory factors. Cerebral microvessels of patients with AD display higher levels of tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, chemokines (CCL2 and IL-8), MMP, and leucocyte adhesion molecules than controls do.63 Evidence suggests that a number of inflammatory mediators can regulate Aβ production (Fig. 3). Zhao et al.64 observed that stimulating astrocytes using such inflammatory cytokines as TNF-α and interferon-γ (INF-γ) increased Aβ secretion and the levels of APP and β-secretase. On the other hand, research has shown that exposure to Aβ promotes a series of proinflammatory responses in brain endothelial cells. Interaction between Aβ and the RAGE receptor regulates CCR5 expression and promotes T cell migration through the BBB.65 Exposure of brain endothelial cells to Aβ in vivo leads to overexpression of such inflammatory genes as MCP-1, IL-1β, and IL-6, as well as increased prostaglandin production.63
These findings point to a molecular connection between vascular dysfunction, neuronal damage, and inflammation in AD. In line with this idea, cerebral microvasculature may be involved in a destructive cycle of events in which inflammation precedes Aβ deposition and in turn, Aβ promotes the release of inflammatory mediators.
Treatment approaches to Alzheimer disease based on the vascular hypothesisGiven current evidence suggesting that cerebral hypoperfusion and inflammation play a major role in AD development, such drugs as non-steroidal anti-inflammatory drugs (NSAID), statins, and hypertensive drugs are considered a promising alternative for the prevention and treatment of AD.58,66
Several retrospective epidemiological studies suggest that NSAIDs significantly reduce the risk of developing AD,67,68 which confirms results from studies in animal models of AD indicating that anti-inflammatory drug therapy reduces Aβ deposition in the brain.69 However, prospective clinical trials of the therapeutic effects of NSAIDs have reported no benefits.70,71 These discrepancies may be due to the fact that epidemiological studies include patients whose clinical manifestations are not yet evident, whereas clinical trials include patients whose symptoms are clinically detectable. Given the wide range of anti-inflammatory drugs and the differences in their pharmacological activity, optimising drug selection in clinical trials is essential. To achieve this aim, we must gain a better understanding of the mechanisms involved in the inflammatory processes contributing to AD progression.72
Clinical trials have failed to show any efficacy of statins for AD prevention and treatment.73–75 In contrast, experimental studies with animal models have provided evidence that statins may prevent senile plaque formation. However, no evidence supports these drugs being able to repair neuronal damage or degrade existing senile plaques.76,77 A plausible hypothesis suggests that clinical trials may have been conducted in patients who had already missed the window of opportunity; these patients may have been treated too late, when damage was already irreversible or there was nothing left to preserve.78
As for antihypertensive treatment, a clinical trial by Forette et al.79 showed that use of antihypertensive drugs was associated with lower incidence of dementia. More specifically, Yasar et al.80 showed that use of diuretics, angiotensin-1 receptor blockers, and angiotensin-converting enzyme inhibitors was associated with a lower risk of developing AD in cognitively healthy patients. In patients with mild cognitive impairment, however, only use of diuretics was associated with lower risk of AD. Whether antihypertensive drugs are suitable for treating AD is still under debate; further clinical trials are necessary to explore the true therapeutic value of these drugs.81
On the other hand, our expanding knowledge of AD pathophysiology and its connection with vascular dysfunction has led to new therapeutic targets being identified. Thanks to the study of RAGE-mediated responses in the development of AD, blocking RAGE is thought to be a promising strategy for AD treatment. Likewise, LRP1 may constitute a new therapeutic target for AD. Soluble LRP1 circulating in plasma binds to plasma Aβ and promotes its degradation. Therefore, LRP1 fragments may have a great therapeutic potential as Aβ clearance agents in AD.82 Clinical data are insufficient to validate the therapeutic potential of these novel strategies.83 Only one recent clinical trial has provided evidence that low doses of a RAGE inhibitor can slow cognitive decline in patients with AD.84 In any case, the vascular hypothesis of AD has provided and will continue to provide insight on new molecular mechanisms that may act as pharmacological targets for AD treatment.
ConclusionsThe vascular hypothesis of AD suggests that a great number of pathogenic pathways having to do with brain blood vessels may be the initial pathogenic stimulus. This idea opposes the traditional concept that AD is neuronal in origin and that the underlying vascular disease is secondary to neurodegeneration. In any case, the available evidence seems to suggest that vascular disease acts synergistically with neurodegenerative changes, which results in greater cognitive decline.
It is difficult to determine whether the vascular component of AD constitutes the cause or rather the effect of the disease. There is no doubt, however, that vascular disease plays a major role in AD progression and that it is associated with neuronal dysfunction. BBB disruption and decreased cerebral blood flow constitute the nexus between vascular risk factors and neurodegeneration. BBB disruption favours Aβ accumulation and decreases the effectiveness of Aβ clearance mechanisms. Decreased cerebral blood flow, on the other hand, increases APP expression and β-secretase activity, which results in greater Aβ accumulation. From this point, cerebral hypoperfusion would favour Aβ accumulation by means of a positive feedback mechanism; Aβ accumulation, in turn, would aggravate vascular dysfunction, causing inflammation and oxidative stress. This would accelerate neurodegeneration and cause the cognitive deficits typical of AD.
Understanding the influence of the vascular system on AD development sheds light on new therapeutic targets and provides a new perspective on AD treatment options. Studying the impact of vascular dysfunction on the pathophysiology of AD is therefore essential.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Rius-Pérez S, Tormos AM, Pérez S, Taléns-Visconti R. Patología vascular: ¿causa o efecto en la enfermedad de Alzheimer? Neurología. 2018;33:112–120.
