


El síndrome de Hadju-Cheney (SHC) está caracterizado por acroostelisis, hiperextensibilidad articular, persistencia de suturas, huesos wormianos, micrognatia y pérdida prematura de los dientes. También presentan talla baja, inteligencia normal, manifestaciones secundarias a impresión basilar, sordera y alteraciones del lenguaje, osteoporosis con tendencia a fracturas vertebrales y de huesos largos. También dolor en manos y pies. Presentamos el caso de una niña de nueve años de edad, con diagnóstico de SHC y calcinosis cutis, una asociación no descrita previamente en la literatura médica.
Hajdu-Cheney Syndrome (HCS) is characterized by acroosteolysis, joint hyper extensibility, sutures persistence, wormian bones, micrognathia and premature loss of teeth, short stature and normal intelligence, neurologic manifestations as paresthesias, secondary to basilar impression, hearing loss and speech alterations, osteoporosis with long bones and vertebrae fractures tendency as well as pain in hands and feet. We present the case of a 9-year-old girl with HCS and calcinosis cutis, an association not previously described in the literature.
Pagina nueva 4
Introduction
Described as acroosteolysis with osteoporosis as well as changes in mandible and skull, also known as Hajdu-Cheney Syndrome, is a rare disease with very few reported cases around the world. The first description of HCS was done in 1948 by Hajdu and Kauntze in a patient with cranial and skeletal dysplasia associated with peripheral dysostosis and spinal osteoporosis. In 1965, Cheney describes a family in Michigan USA, with a disorder of connective tissue manifested as acroosteolysis, wormian bones and mandible hypoplasia; at the same time, describes the autosomal dominant inheritance with expression variability and some other sporadic cases.1,2 Hajdu-Cheney syndrome is caused by heterozygous mutation in the NOTCH2 gene (MIM 600275) on chromosome 1p13-p11.
Recently, truncating mutations in exon 34 of the NOTCH2 gene have been identified. All the mutations occurred in exon 34, the last exon of the NOTCH2 gene, and all were predicted to lead to truncation of the protein product before complete translation of the PEST domain, which mediates proteosomal destruction of the protein. The no-sense mutations were not taken into account due to corrupt mRNA, and the truncated proteins were expressed. Thus, the mutations resulted in persistence of the intracellular NOTCH signal, consistent with a gain of function.3
Case report
The present case is a 9-year-old female from Mexico City, product of the first pregnancy, with complications at the sixth gestational month and resolved by cesarean section at 40 weeks, birth weight 3 025 Kg, length 48 cm, Apgar score 8/9.
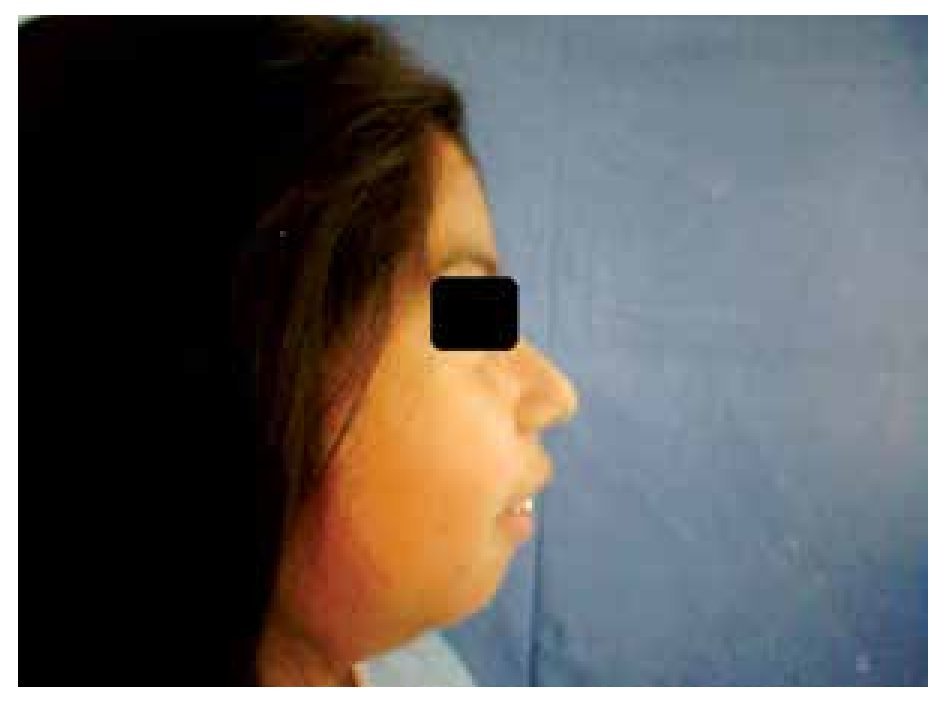
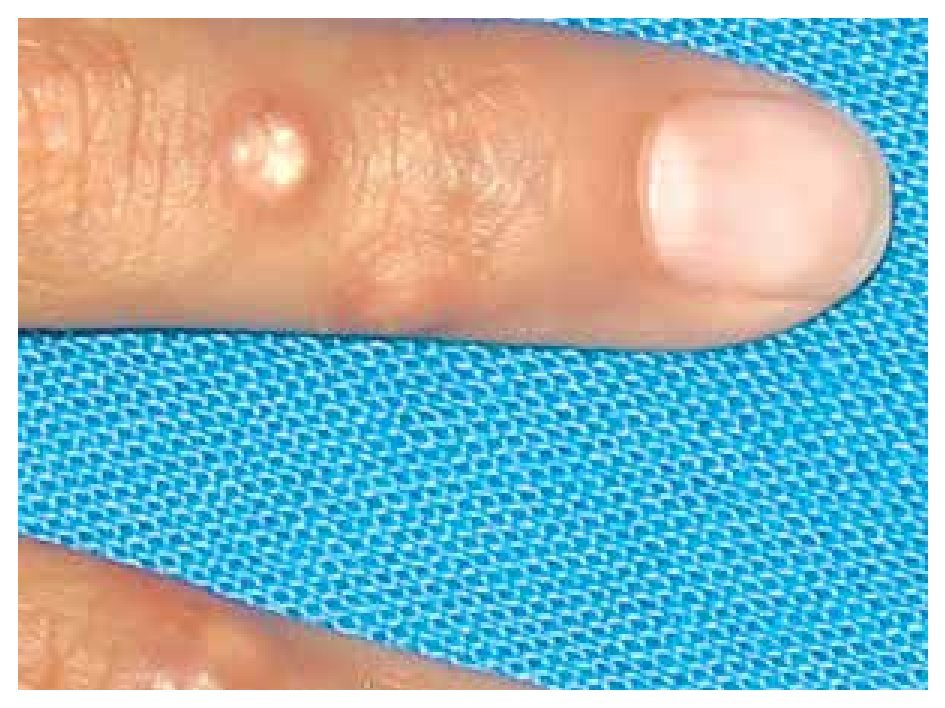
When she was four years old, parents notice spots in upper lids, elbows, knees as well as hard nodules, white or yellowish, initially remittent with eritematous and painful surrounding zones, located mainly in hands and feet. The physical examination revealed a weight of 27.5 Kg, length of 121.5 cm, head circumference of 48 cm. She presents hyperchromic and achromic lesions in upper lids. Micrognathia, high palate, brown au lait spots in thorax and abdomen regions (Figure 1). Also, she had limbs with hyperextensible joints, clusters of hyperchromic and achromic spots in elbows, knees, and poplitean regions; white-yellowish tumoral formations approximately 0.3-0.5 cm in length, with very hard consistency on hands, feet and mainly on fingers (Figure 2), there are lineal, irregular brown au lait spots of 3 cm in length.
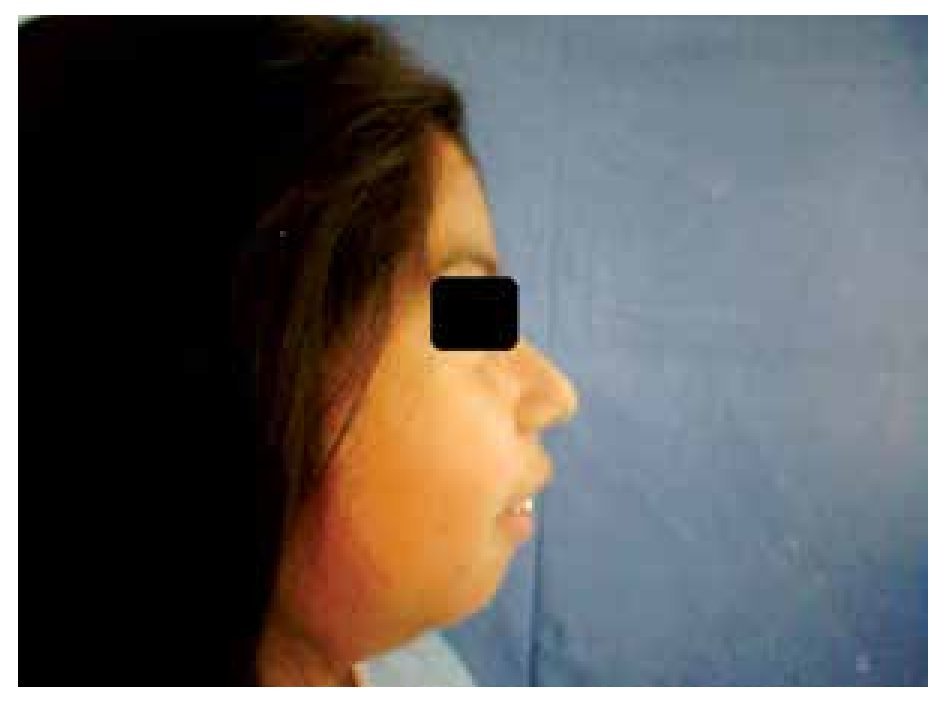
Figure 1. Retrognathia.
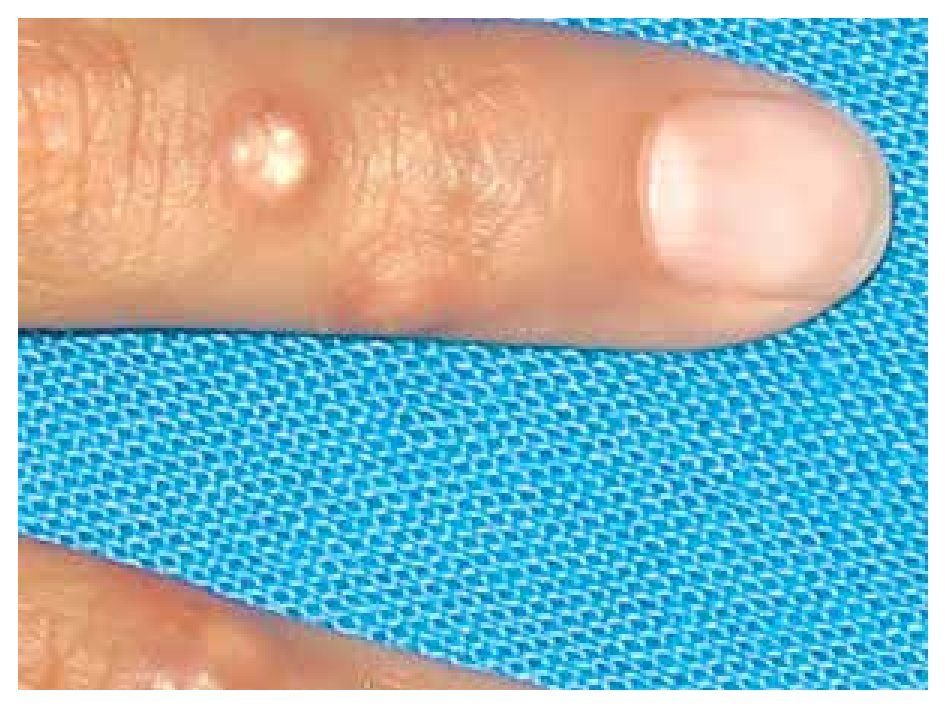
Figure 2. Calcinosis cutis lessions in fingers.
Complete blood count, reticulocytes, BUN, creatinine, glucose, basic metabolic panel and hepatic function test all in normal ranges.
Calcium: 10 mg/dl, phosphorus: 4.6 mg/dl and magnesium: 2.1 mg/dl.
Urine sample analyses with amorphous phosphates limited, phosphorus depuration 23.81 (normal < 2-8 ml/ min). Phosphorus tubular reabsorption: 15.17 (normal > 85%). Parathyroid hormone values: 4.58 pmol/l (normal 1.6-7).
Cytogenetic analysis: 46, XX without numeric or structural alterations.
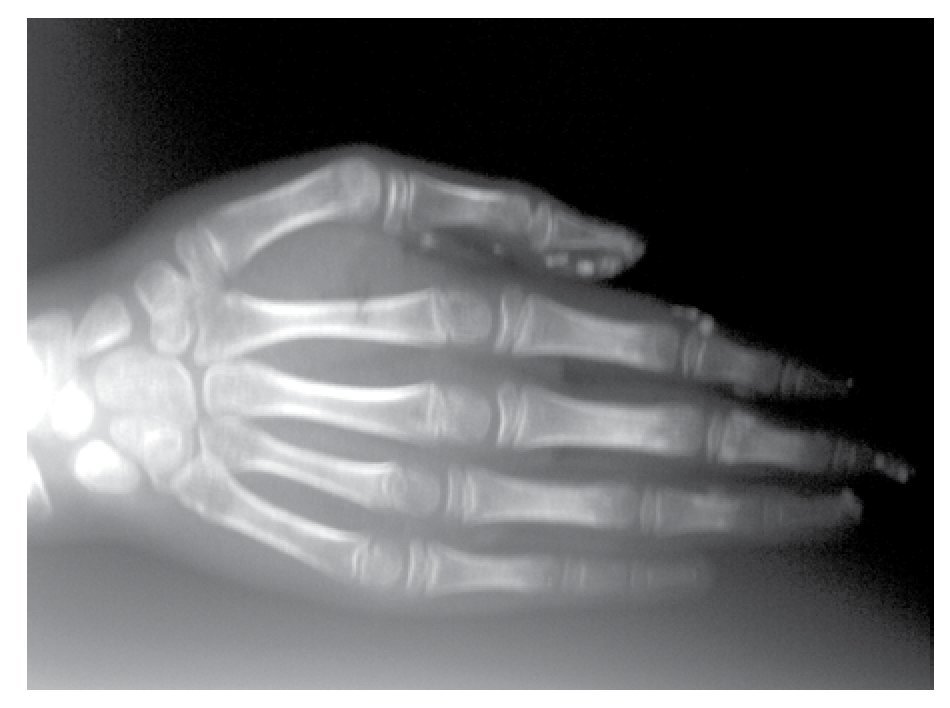
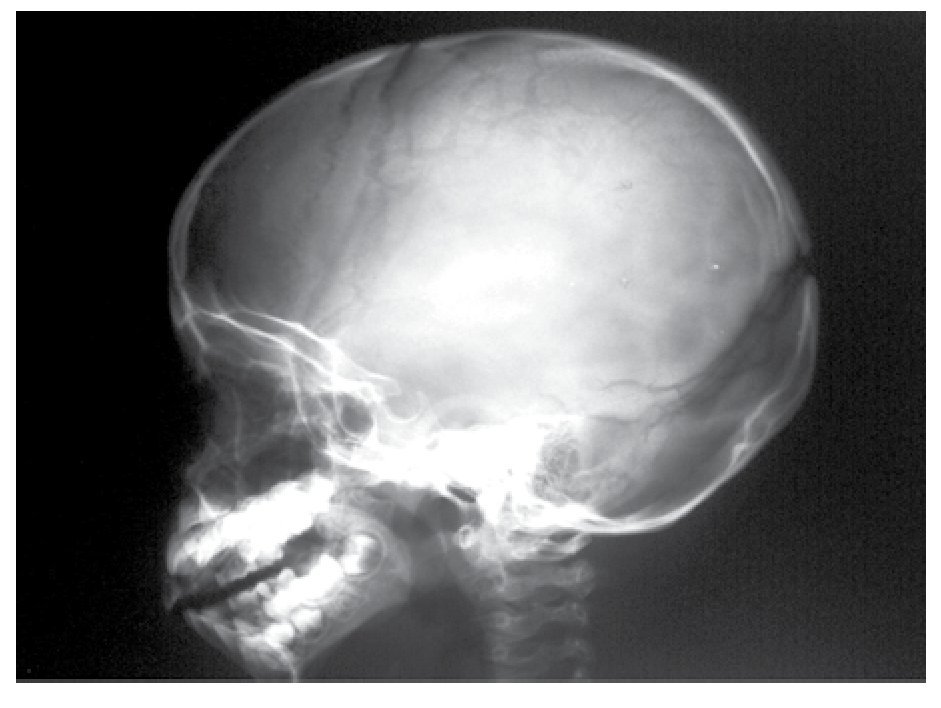
AP and lateral radiological studies of the cranium revealed wormian bones and open sutures. Hand X-ray studies demonstrated subcutaneous calcifications (Figure 3). Simple and contrasted CAT studies were reported as normal. Renal gammagram with proper renal function but dilation of urinary tracts secondary to partial obstruction of urine flow which improves under diuretic administration. Abdominal CAT reported as normal with no calcifications in kidney or gallbladder.
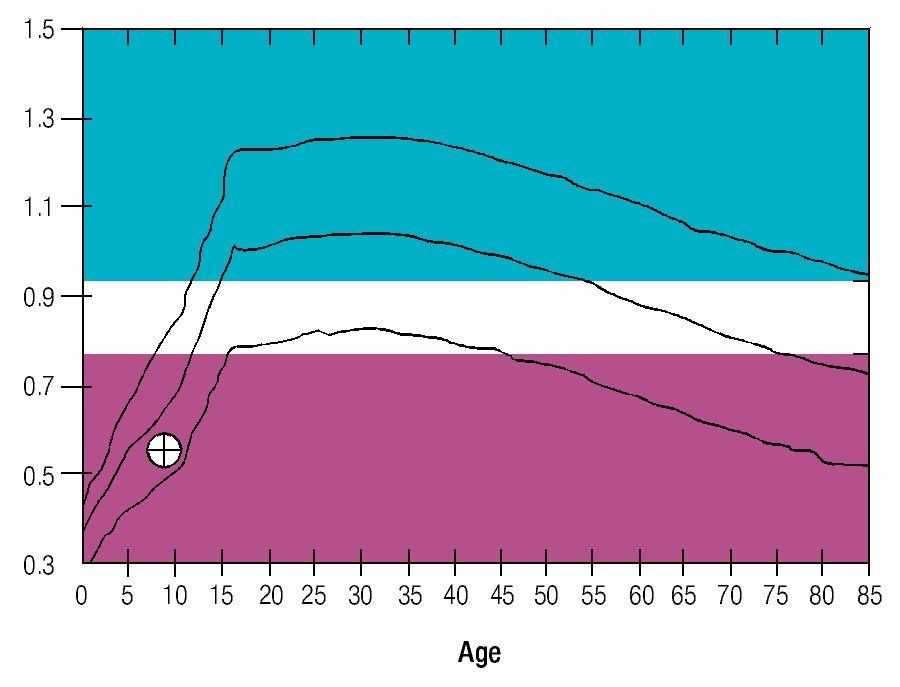
Figure 3. Subcutaneous calcification in hand and osseous resorption on distal phalanges.
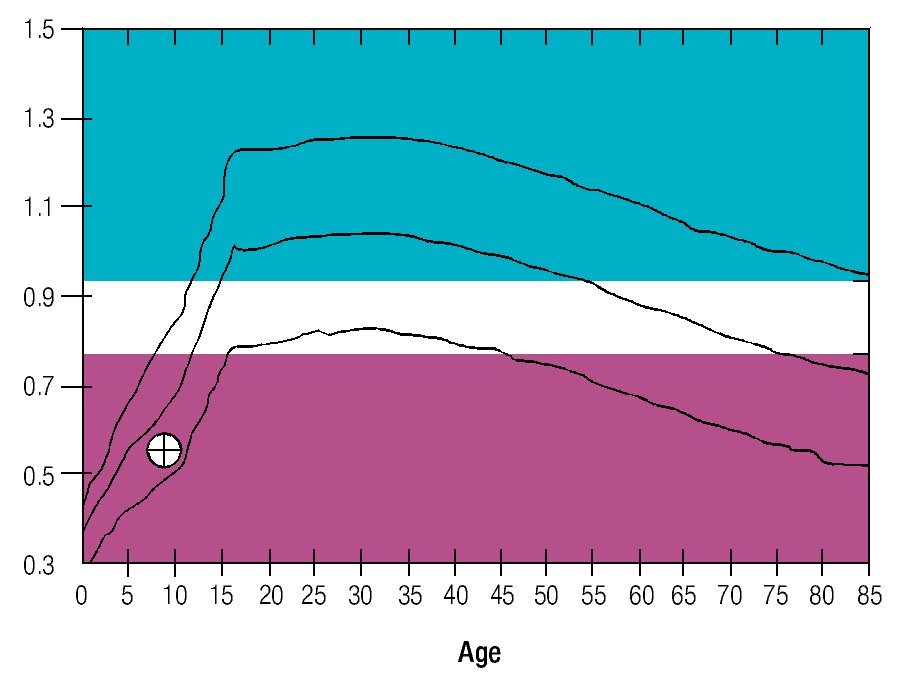
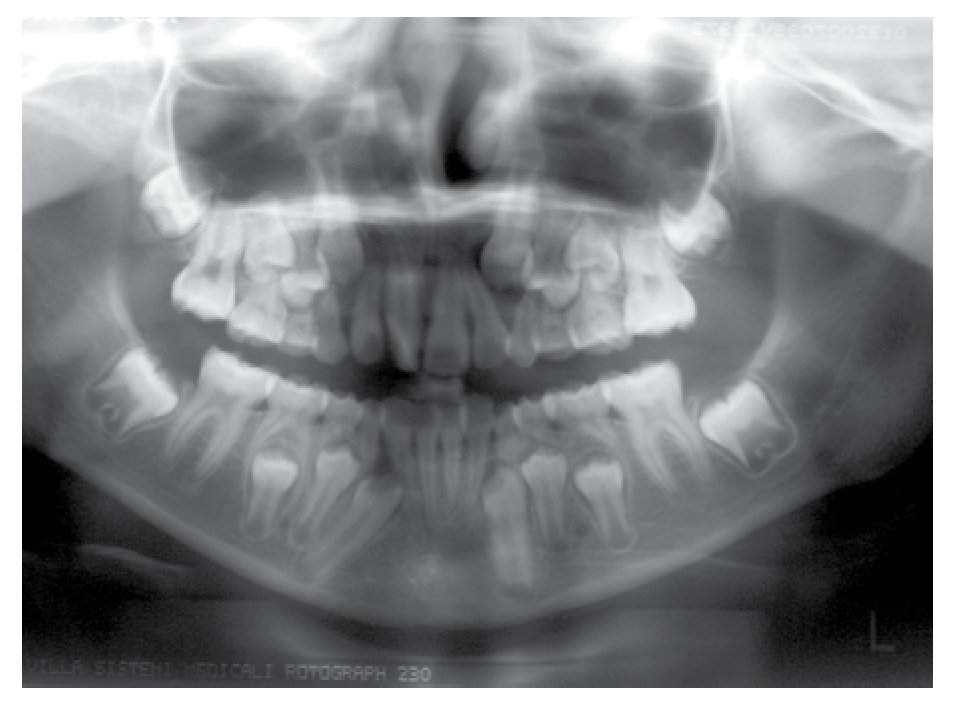
Osseous densitometry shows an important sub mineralization (Figure 4). Orthopanthography classification as Prudzansky 1 (Figure 5). Evaluation by orthodoncists revealed Angle class II malocclusion, micrognathia, retropositionated mandible, short mandible branches, very small mandible condyles and bizarre dental clustering (Figure 6).
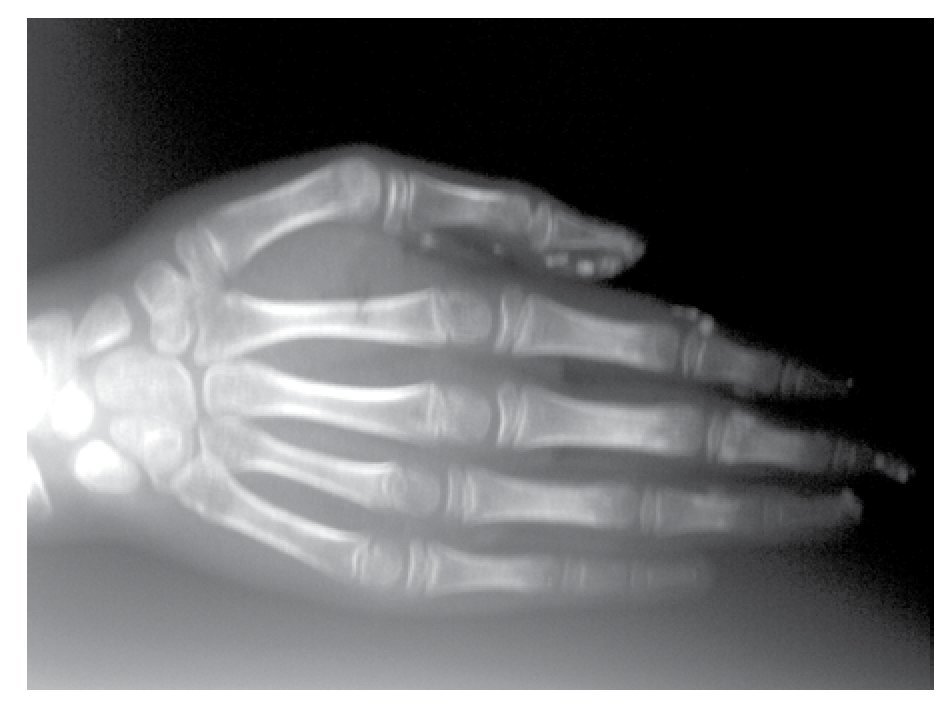
Figure 4. Densitometric study showing vertebral column osteoporosis.
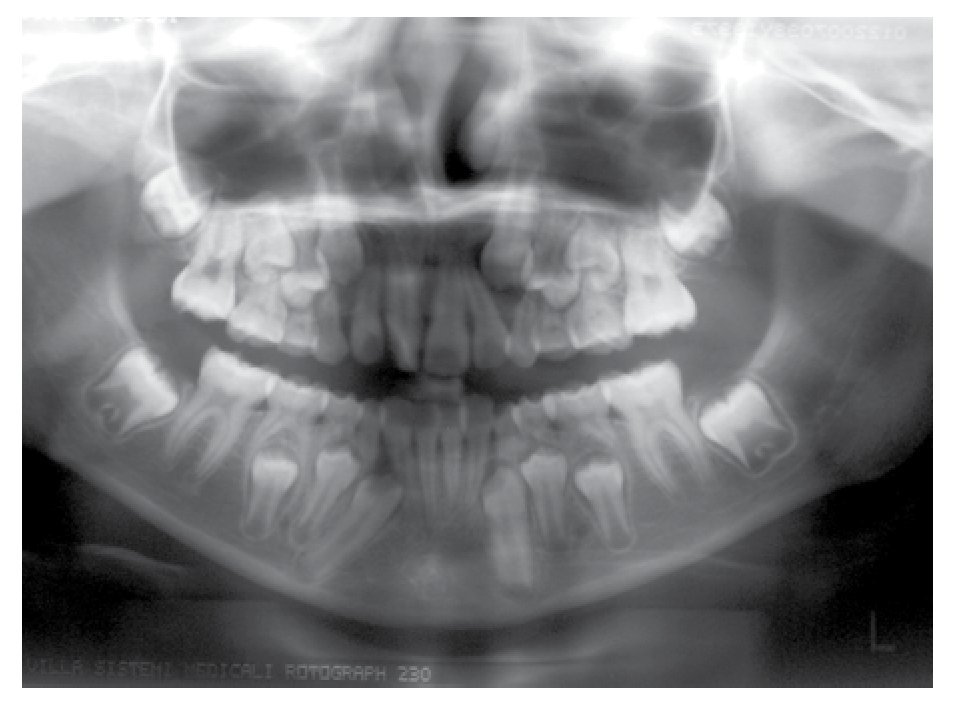
Figure 5. Orthopantomography showing hypoplasic condiles.
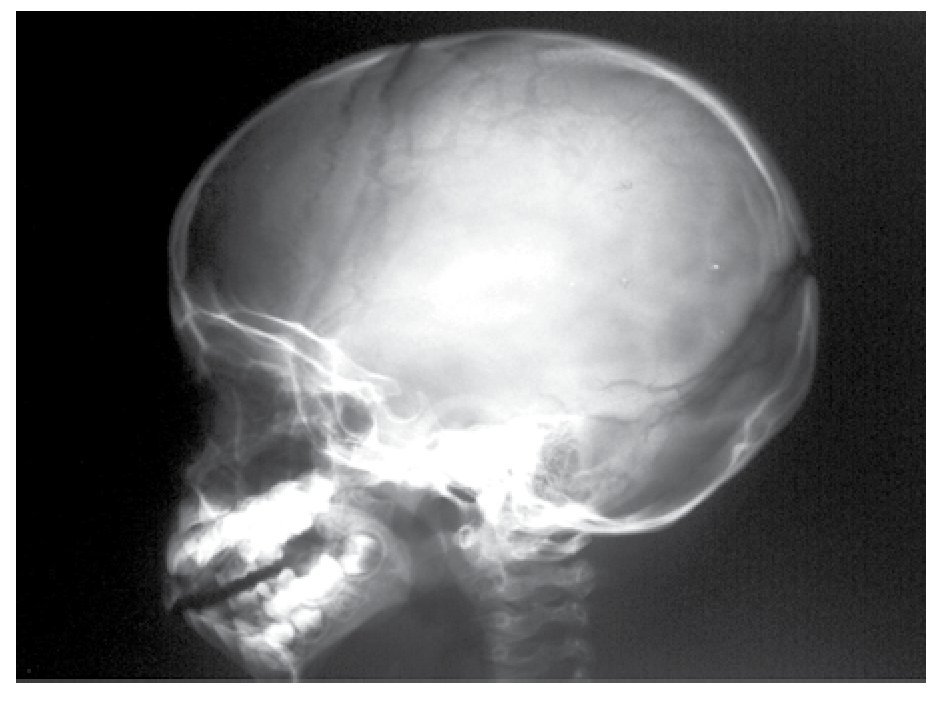
Figure 6. lateral view of skull showing wormian bones and open sutures.
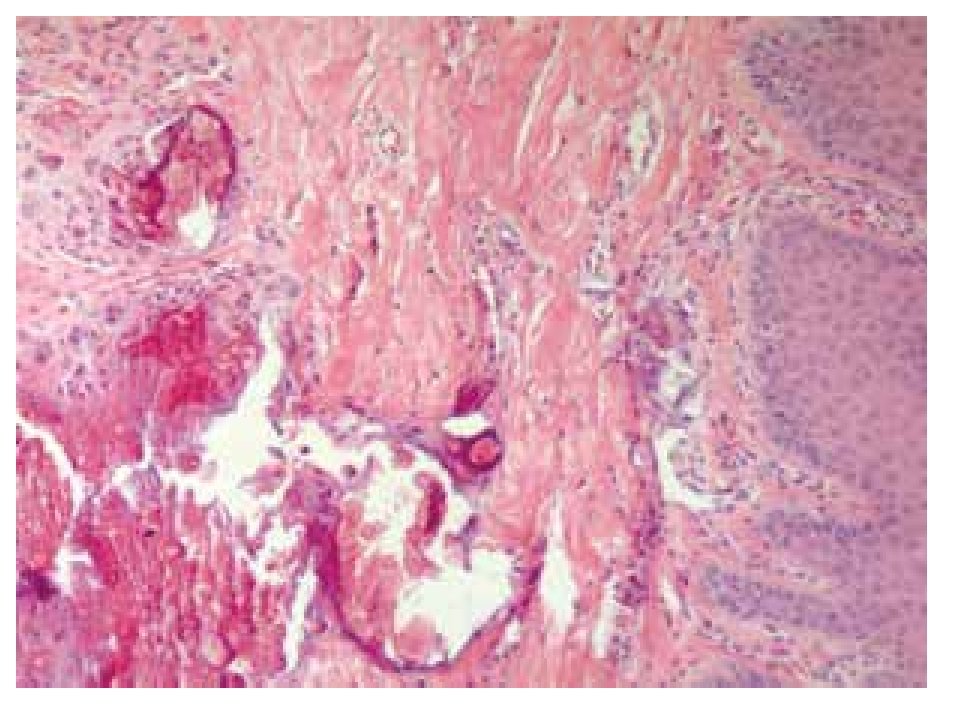
Skin biopsy reported numerous calcifications in papillary and reticular dermis as well as in epidermis sections, findings suggestive of calcinosis cutis (Figure 7). Skin also exhibited dystrophic and irregular calcium deposits, surrounded by mononuclear cells, mainly histiocytes in both epidermis and dermis layers.
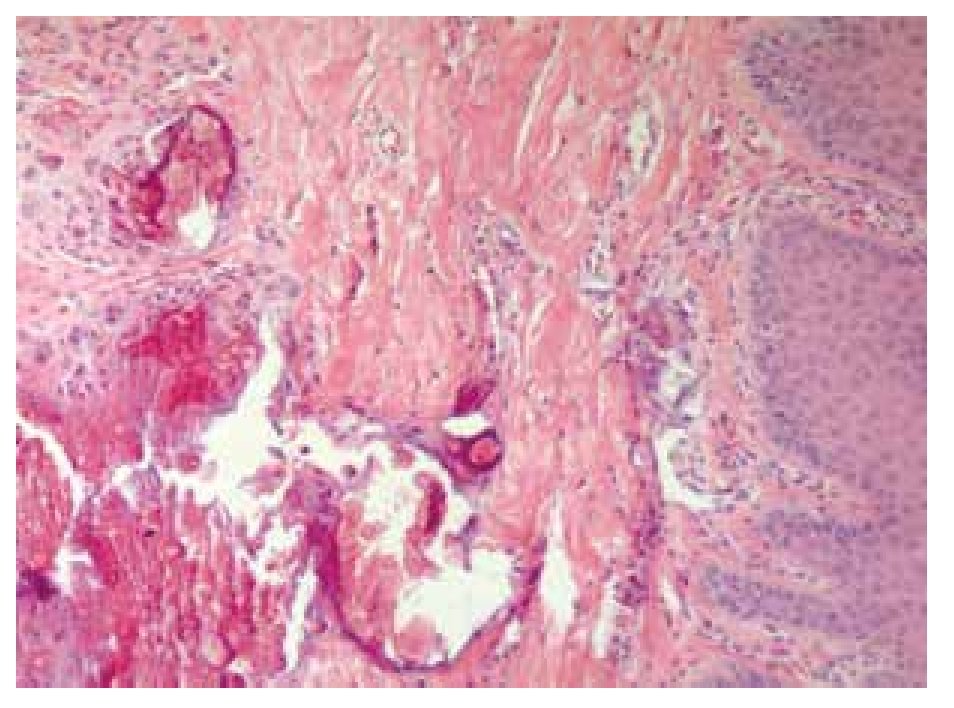
Figure 7. Dermis biopsy with calcium deposits.
Discussion
Diverse clinical manifestations are associated to HCS, among those are osteoporosis, multiple skull, spine and fingers fractures, joint hyperextensibility, short stature, persistence of cranial sutures, early loss of teeth, progressive loss of auditive acuity and, less frequently, overgrowth of sella turca without endocrine changes, progressive loss of bone fragments in phalanges, local neurovascular dysfunction and renal cysts among other findings.2
The first case of HCS in a male patient was reported in a 31-year-old male with serious respiratory problems, nasal voice and nocturnal hypoventilation with episodes of Cheyne-Stokes respiration, progressing to laryngeal noise and vocal cord paralysis as well as diminished conduction speed of superior laryngeal nerve.
Neurologically HCS presents hydrocephalus, trigeminal neuralgia and basilar impression with central nervous system (CNS) compromise, which altogether with the unknown etiology and pathogenesis, represents a bad prognosis. Tanimoto et al, described a HCS case in a 41-year-old patient with neurological signs associated with cervical siryngomielia, surgically resolved by 1st cervical laminectomy and posterior occipito-cervical fusion.4
Another HCS case in a 48-year-old patient with progressive basilar invagination, generating a bilateral visual loss; MRI demonstrated a massive overgrowth of both intraortbitary optic nerves.5 Fryns et al confirmed the progressive nature of neurological symptoms in patients with HCS.6
Acroosteolysis is described as a secondary feature to the metabolic disorder involved in the progressive destruction of distal phalanges in HCS.7 A very rare clinical case with distal osteolysis, beside cervical osteolysis requiring surgical management for cervical instability.8
Diffuse osteoporosis in HCS is secondary to reduction in bone formation and an increment of its resorption, leading to distal osteolysis and osteoporosis. Histological studies in biopsies demonstrated severe osteoporosis with important reduction in the formation of bone and calcium kinetics studies showed a 2.8 times increment in osteoclastic resorption. Serum PTH, 1, 25 (OH)2 D, alkaline phosphatase, were normal but some biochemical studies demonstrated high levels of acid phosphatase.9 Above mentioned data suggest that HCS and osteoporosis could result from the same inductor mechanism as in distal osteolysis and could be substituted with local activation of osteoclastic resorption factor10 and has been suggested suggesting that the former is related with insufficient bone formation and increase on its resorption.11
Kidney defects such as renal cysts, vesicoureteral reflux, glomerulonephritis and secondary hypertension have been found as frequent alterations in HCS, beside bilateral renal hypoplasia with chronic renal failure.12
Siklar et al, described a case of a 9-year-old boy with physical and radiological features of HCS, associated with growth hormone deficiency and peripheral neuropathy.13
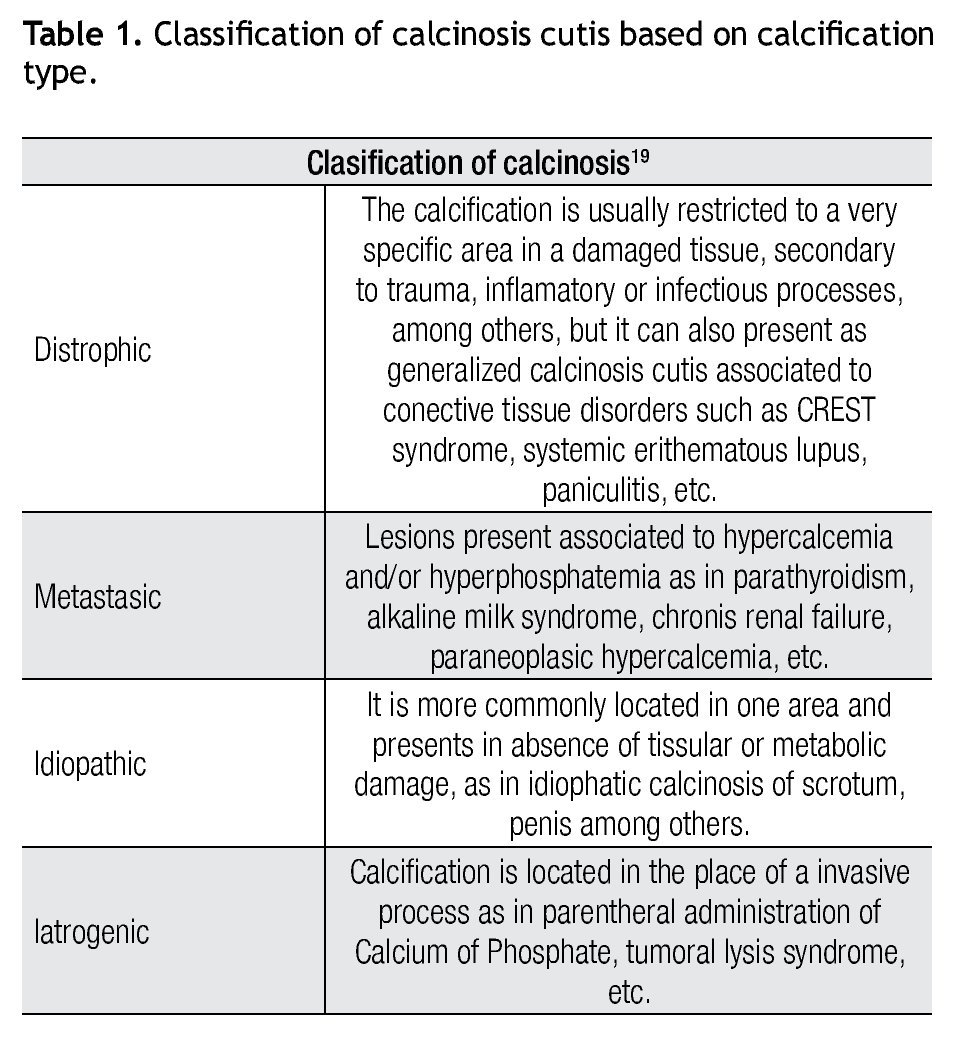
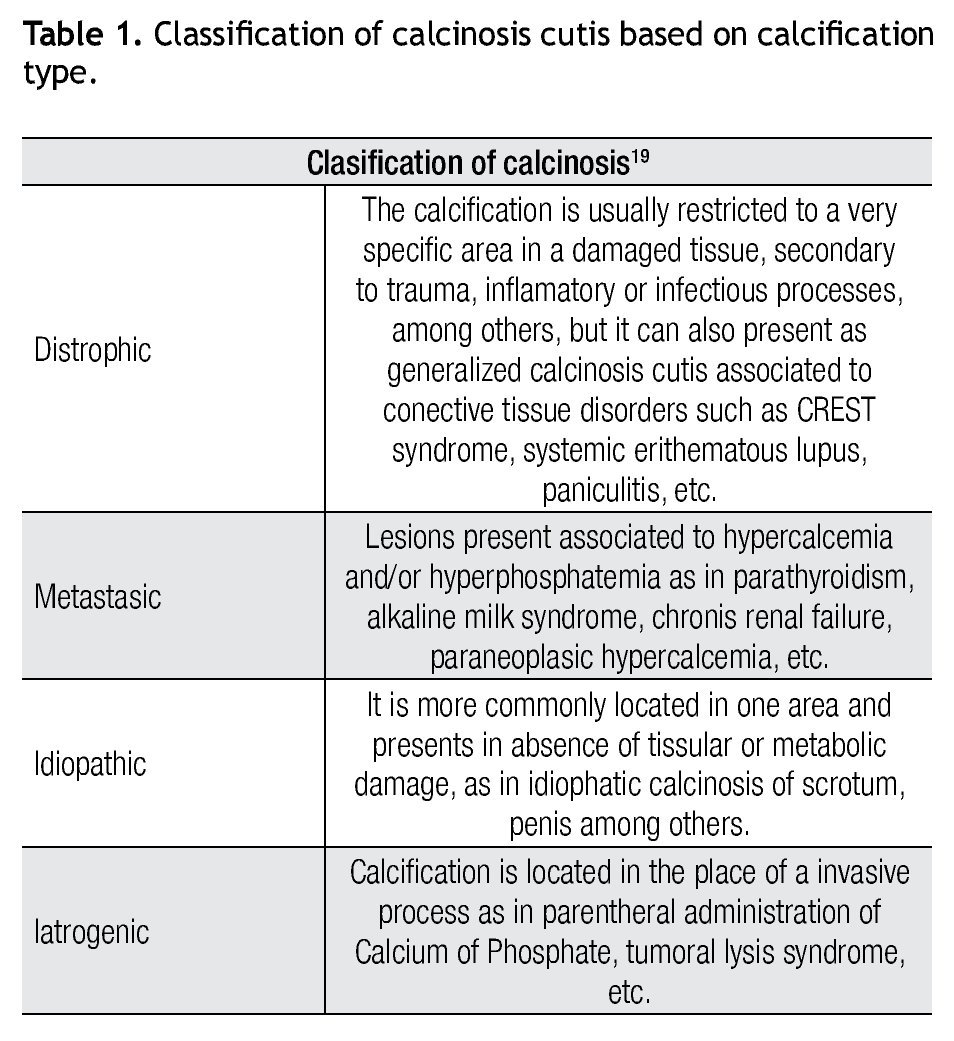
Calcinosis cutis was first described in 1855 by Virchow and is defined as insoluble calcium deposits in cutaneous tissues. It is classified in different types (Table 1). Calcium is a very important element in biological processes, promoting keratinocytes proliferation on skin, differentiation and cell adhesion, and influence on epidermal cell growth. In calcinosis, calcium and phosphorous deposits precipitate to form an amorfous material characteristic of this pathology.
The most frequently described type of calcinosis cutis is the dystrophic, on which most of the lesions develop gradually and is asymptomatic, with a benign course and no gender predilection. The subepidermal calcified nodules are more frequent in children being calcinosis cutis localized in them, and universal types more frequent in the second decade of life. Tumoral calcinosis, the most severe type, appears during the first or second decade, being more frequent in black population. The clinical features of calcinosis cutis vary depending if it is settled on a normal or damaged tissue. The most frequent malformation is a whitish subcutaneous neoformation, single or multiple, petreous consistency with micro-ulcers or erythema over the surface or around the lesions (Figure 2).
The idiophatic form of calcinosis cutis has been associated with four different and well recognized syndromes such as: fibrodysplasia ossificans progressiva, progressive osseous heteroplasia, cutaneous osteoma plate shape and Albright hereditary osteodystrophy.14 Idiopathic types describes calcified subepidermal nodules in a single lesion from 3-11 mm in exposed areas on the head and extremities congenital or aquired.15
Microscopically, all cases of calcinosis cutis, displayed calcium salts deposits with amorphous shape and variable in size inside dermis or subcutaneous cell tissue, surrounded by granulomatous inflammatory infiltra- tes. Calcifications are mainly formed by hydroxiapatite and calcium phosphate precipitated crystals, favored by rises in calcium and phosphate serum levels even without a previous tissue lesion.15
Some drugs have been used for the treatment of calcinosis cutis, most of them directed toward lowering of calcium deposits in skin. Among those drugs are probenecid, potassium PABA acid and warfarin which at low doses reduces the gamma carboxiglutamic acid synthesis, a vitamin K dependent protein involved in calcification process. Among anti-inflammatory drugs prescribed for the disease, are colchicine, steroids, allendronate, disodic etidronate and diltiazem. As a last choice, the surgical remotion of calcium deposits should be considered if masses are painful, become infected recurrently, they present ulcerations, or present with functional or cosmetic impairment. Some cases of spontaneous regression have been reported.16
The NOTCH2 gene encodes a single pass transmem- brane protein belonging to an evolutionarily conser- ved NOTCH receptor family. NOTCH signaling is activated through cell-cell contact: ligand binding induces cleavage of NOTCH and translocation of the intracellular domain to the nucleus where it regulates gene expression in association with transcriptional cofactors.
The NOTCH receptor expression has been characterized in the developing heart and liver of mice. In the developing mouse heart, both NOTCH1 and NOTCH2 are expressed in the outflow tracts and the epicardium, and in specific cell populations previously shown to express Jag1. These cells are destined to undergo transformation from epithelial to mesenchymal cells. In the newborn mouse liver, NOTCH2 and NOTCH3 are expressed in opposing cell populations, suggesting they play different roles in cell fate determination during bile duct development. Jag1 is also expressed in cells adjacent to those expressing NOTCH2, suggesting a possible ligand-receptor interaction.17
Mitsiadis et al determined that NOTCH2 is involved in tooth development. During early stages, NOTCH2 was expressed in the epithelium and, at more advanced stages of development, it was expressed in the enamel-producing ameloblasts. NOTCH2 was not expressed in the pulp of adult intact teeth, but was reexpressed during dentin repair in odontoblasts and in subdontoblastic cells. TGFB1, which stimulated odontoblast differentiation and hard tissue formation after dental injury, downregulated NOTCH2 expressed in cultured human dental slices.18
We report the case of a prepubertal girl, with features suggesting Hajdu-Cheney Syndrome associated to calcinosis cutis without previous report of this association on the literature.
It would be interesting to search for mutations in NOTCH2 in our patient, in order to confirm if calcinosis cutis it is part of the same disease spectrum and if some polymorphic variant is related. It is suggested that when the physician faces a non classical picture for HCS, it is mandatory to perform a complete battery of studies for mineral determination.
Handling a patient with HCS implies the participation of a multidisciplinary team composed of a medical gene-ticist, a pediatrician, a plastic surgeon, a maxillofacial surgeon, a pathologist among others.
Medical treatment for a HCS patient, includes, alendronate, vitamin C and calcium, all of which improve the osseous calcification with a more favorable prognosis. However, due to the very few cases of the disease, the management experience is limited and more studies are necessary in order to recommend a treatment for HCS associated to calcinosis cutis.
At present, our patient has a conservative management by the Plastic Surgery Service to improve the osseous calcification status, because there are doubts about the ossification of mandible fracture as part of her functional and aesthetic treatment.
Alendronate treatment was initiated with three main objectives: 1) to improve the osseous density to reduce the fracture risk, 2) to favour the calcium kinetics in calcinosis cutis by inhibition of osseous reabsorption and consequently the calcium deposits and 3) to inhibit the production of proinflamatory cytokines IL1, IL6, and TNF with an important reduction of inflamatory process in calcified areas. No specific treatment exist for HCS, but symptomatic relief is the choice.
Conflict of Interest Statement
The authors declare that there is no conflict of interest.
Financial Support
There was no financial support involved in relation to this article.
Corresponding author:
Gerardo R. Zaragoza-Arévalo, M.D.
Centro Médico Nacional "20 de Noviembre", ISSSTE.
Félix Cuevas N° 540, Colonia Del Valle, Delegación Benito Juárez,
Z.P. 03100, Mexico City, Mexico.
Phone: 5200 5003, ext. 14329.
E-mail: gerardozaragoza69@yahoo.com.mx
Received: November 2011.
Accepted: November 2012