




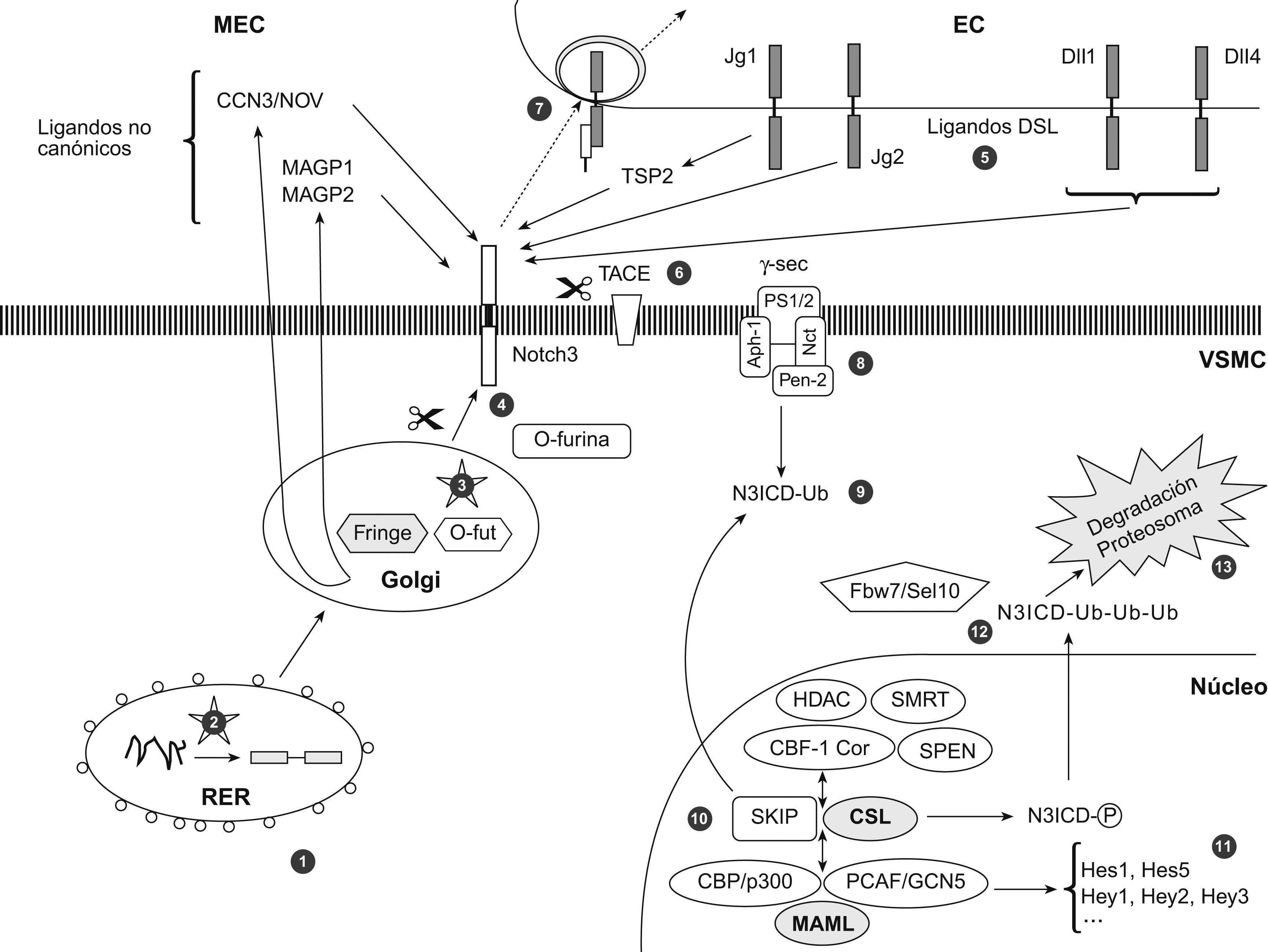
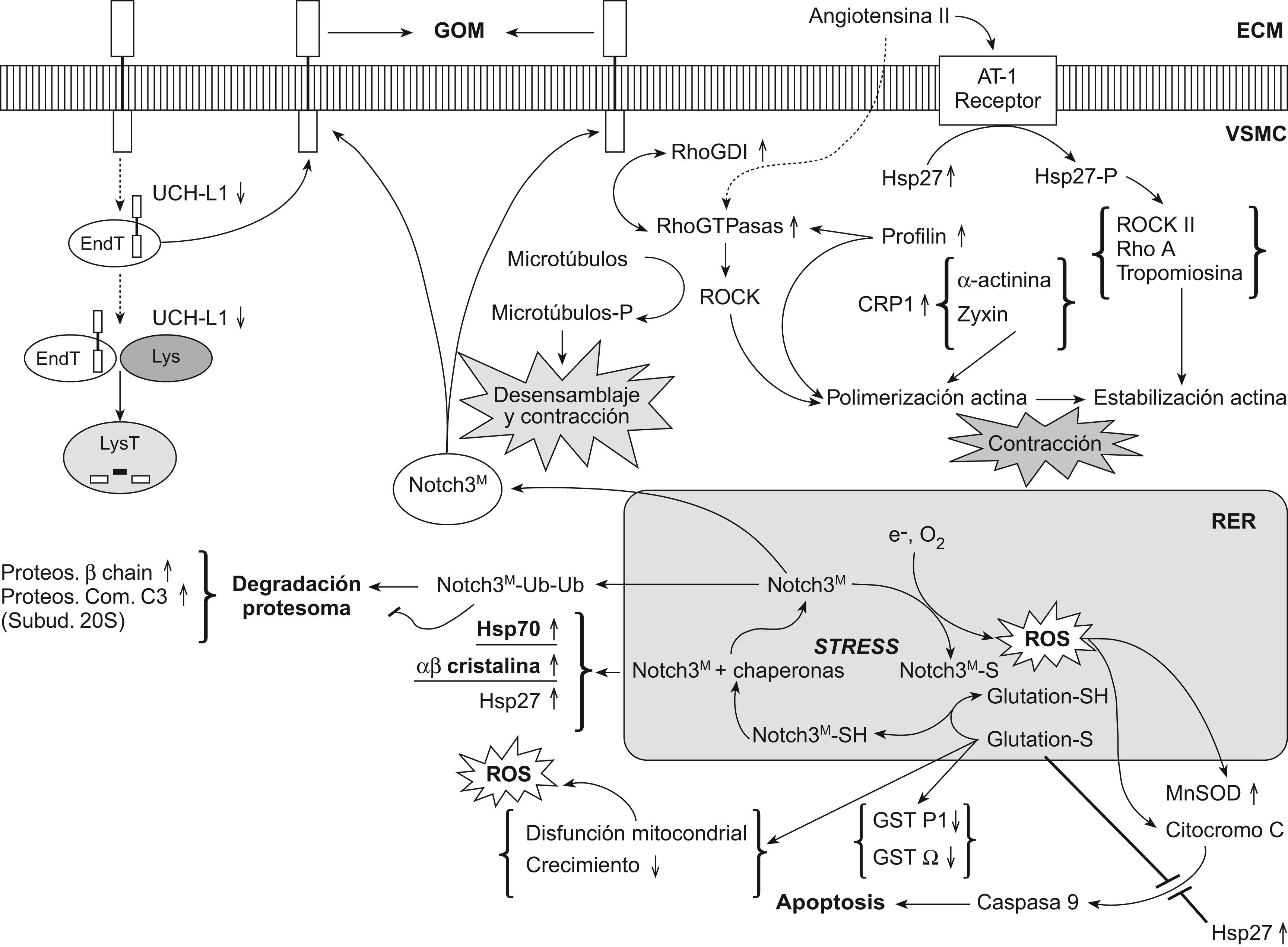
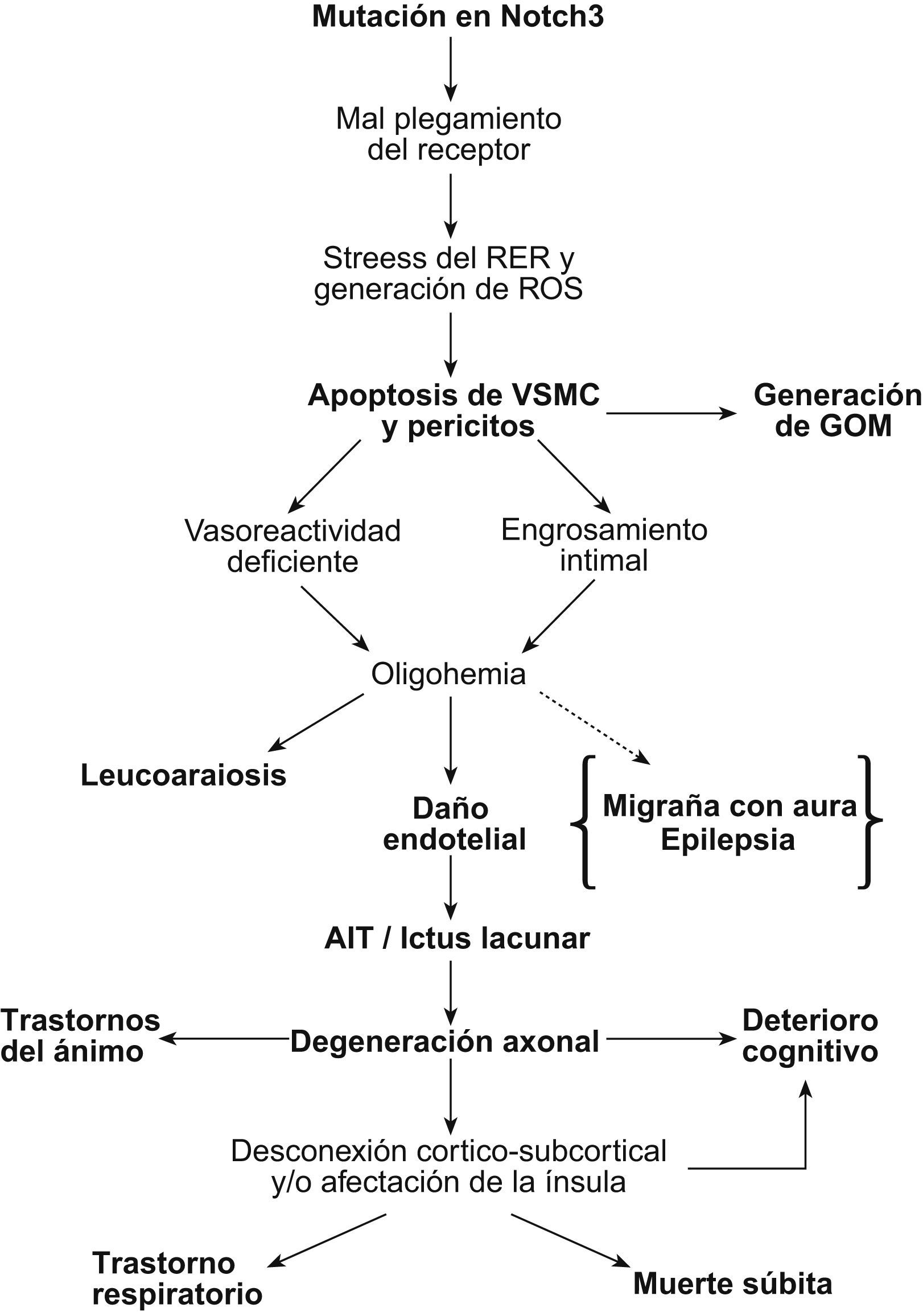
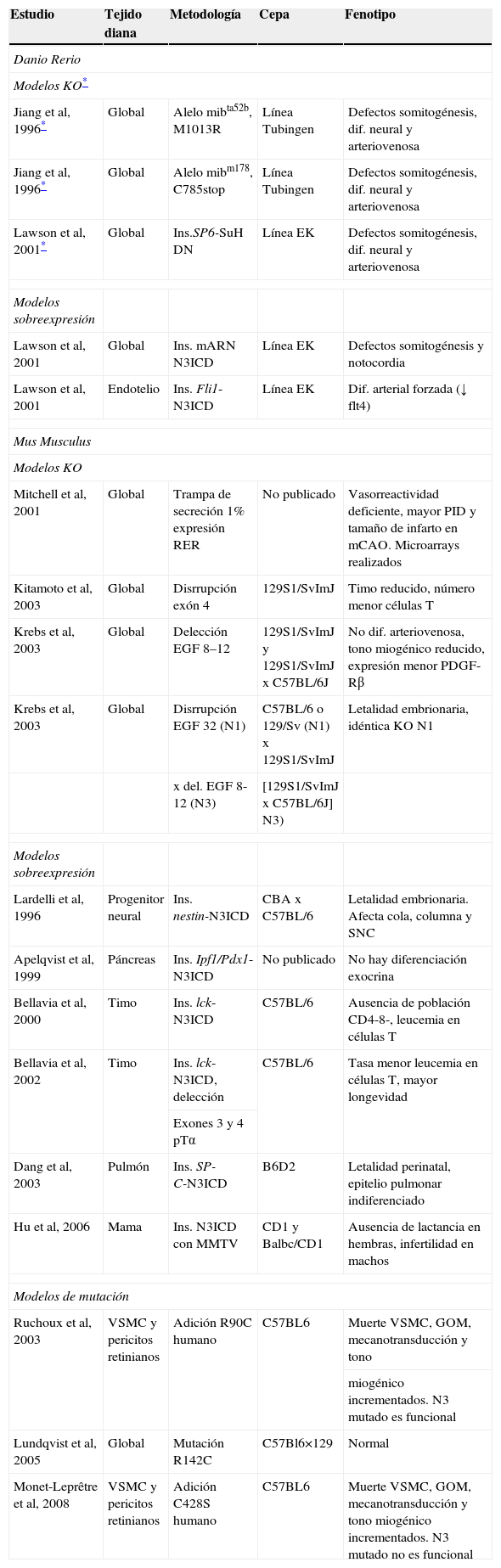
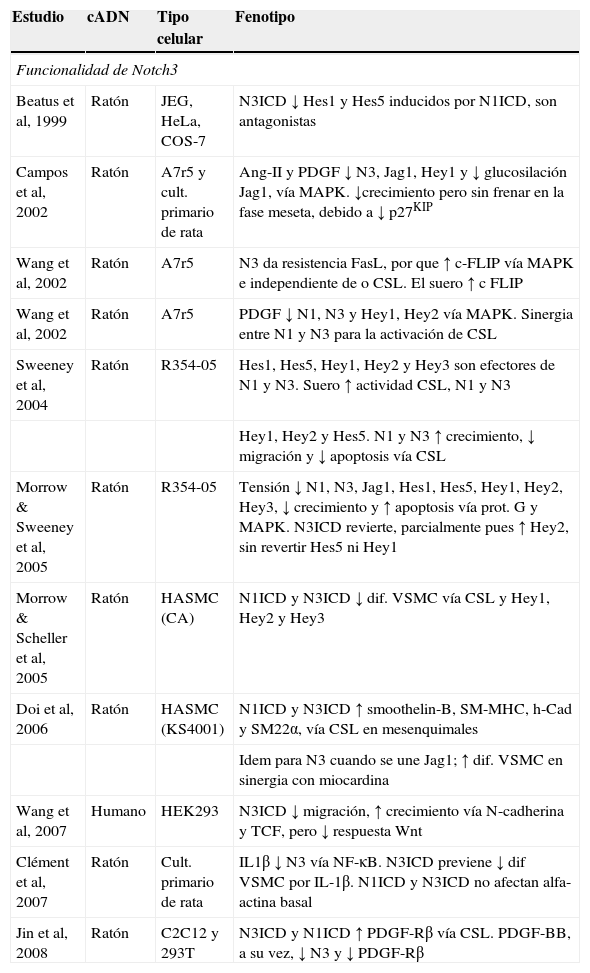
El mecanismo patogénico de la cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL, ‘arteriopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía’) todavía no se conoce, aunque la enfermedad está bien caracterizada a nivel clínico, histológico y genético. La conservación de la vía de Notch en la evolución ha permitido el desarrollo de numerosos modelos animales y celulares para su estudio. En esta revisión se discutirá la validez de los 7 modelos patogénicos propuestos para CADASIL: origen autoinmunitario, afectación mitocondrial, disfunción en la producción de elastina, problemas en la glucosilación del receptor, pérdida de función de NOTCH3, toxicidad de los gránulos osmiófilos y activación prolongada de la respuesta al mal plegamiento proteico. Se tratará, también, la relación entre la degeneración de las células musculares lisas vasculares, las lesiones isquémicas y la sintomatología. Por último, se apuntarán las diferentes teorías para explicar por qué la expresión clínica se circunscribe al sistema nervioso si se trata de una arteriopatía sistémica.
The pathogenic mechanism underlying Cerebral Autosomal Dominant Artheriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) remains elusive although the disease is well characterized at clinical, histological and genetic level. The conservation of the Notch pathway among species allowed the development of several animal and cellular models in order to study it. This review analyzes the reliability of the 7 pathogenic models raised for CADASIL disease: autoimmune origin, mitochondrial dysfunction, loss of Notch3 function, granular osmiophilic material (GOM) toxicity and long term unfolded protein response (UPR) activation. Besides, the relationship between vascular smooth muscle cells (VSMC) degeneration, ischemic lesions and symptoms are discussed. Lastly, some theories are pointed that would explain the exclusiveness of clinical expression to the neural system, being in fact a systemic artheriopathy.

