




Von Hippel-Lindau (VHL) disease is a rare hereditary syndrome that genetically predisposes the affected individuals to develop tumours in multiple organ systems.1,2
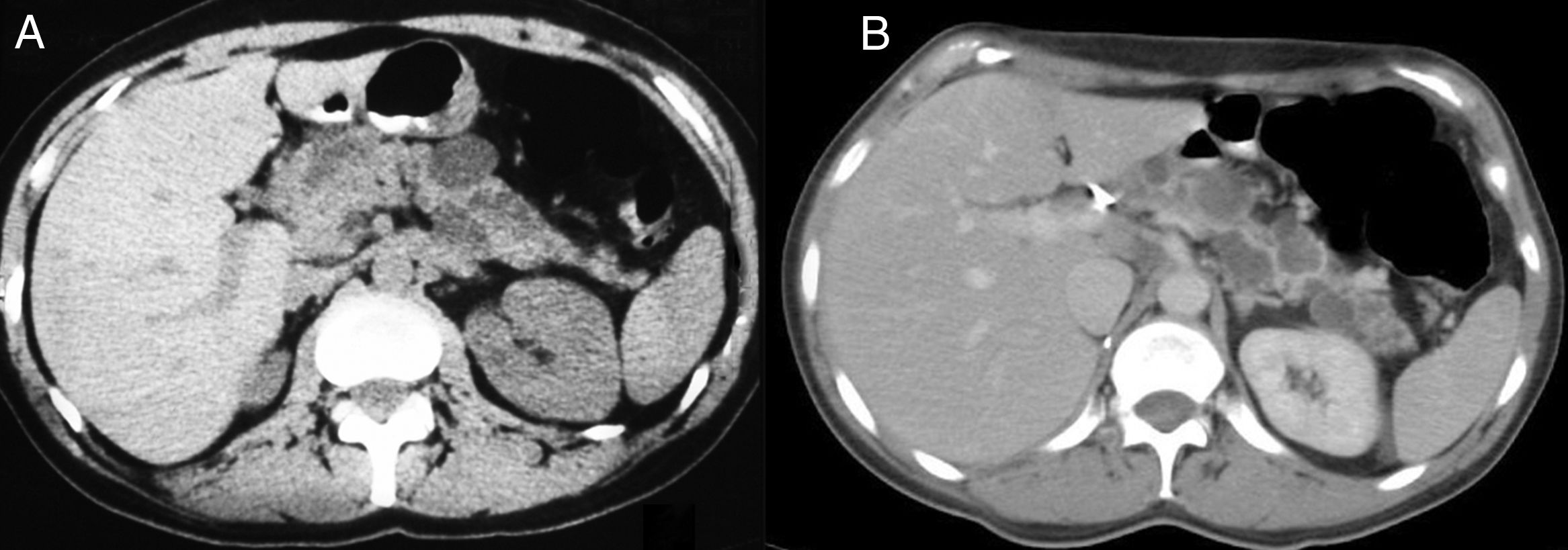
A 43-year-old woman underwent surgery for a right suprarenal pheochromocytoma in 1995. She was undergoing regular monitoring for multiple simple cysts in the pancreatic tail. In 2007, a solid lesion with a 5cm diameter was observed in the head of the pancreas, obstructing the duct of Wirsung, along with cysts in the pancreatic tail (Fig. 1A). Her blood tests showed no significant alterations. A cephalic duodenopancreatectomy was performed, revealing said mass in the head of the pancreas and multiple cysts in the pancreatic parenchyma of the body and tail. Post-surgery, there were no complications. During the histological study, the mass was reported to be a pancreatic neuroendocrine tumour (PNET), testing positive for keratins (AE1/AE3), vimentin, synaptophysin and neuron-specific enolase, with a proliferative index of 18% (MIB-1) and 11 mitoses per 10 high-power fields. Also identified were a serous cystadenoma (SCA) and simple pancreatic cysts in the non-neoplastic parenchyma ( Fig. 2A and B). In light of suspected VHL disease, a genetic study was performed and revealed VHL exon deletion, thereby confirming the disease. Magnetic resonance imaging (MRI) of the head ruled out disorders of the hypothalamic-pituitary region. After 10 years of monitoring, the patient presented a pT1a stage clear renal cell carcinoma which was excised by means of a right nephrectomy. The cysts in the pancreatic body and tail showed no significant changes (Fig. 1B) and the patient presented with neither endocrine nor exocrine pancreatic insufficiency.
 Solid lesion with a 5cm diameter in the head of the pancreas, obstructing the duct of Wirsung, along with cysts in the pancreatic tail. (B) Cysts in the pancreatic tail.")
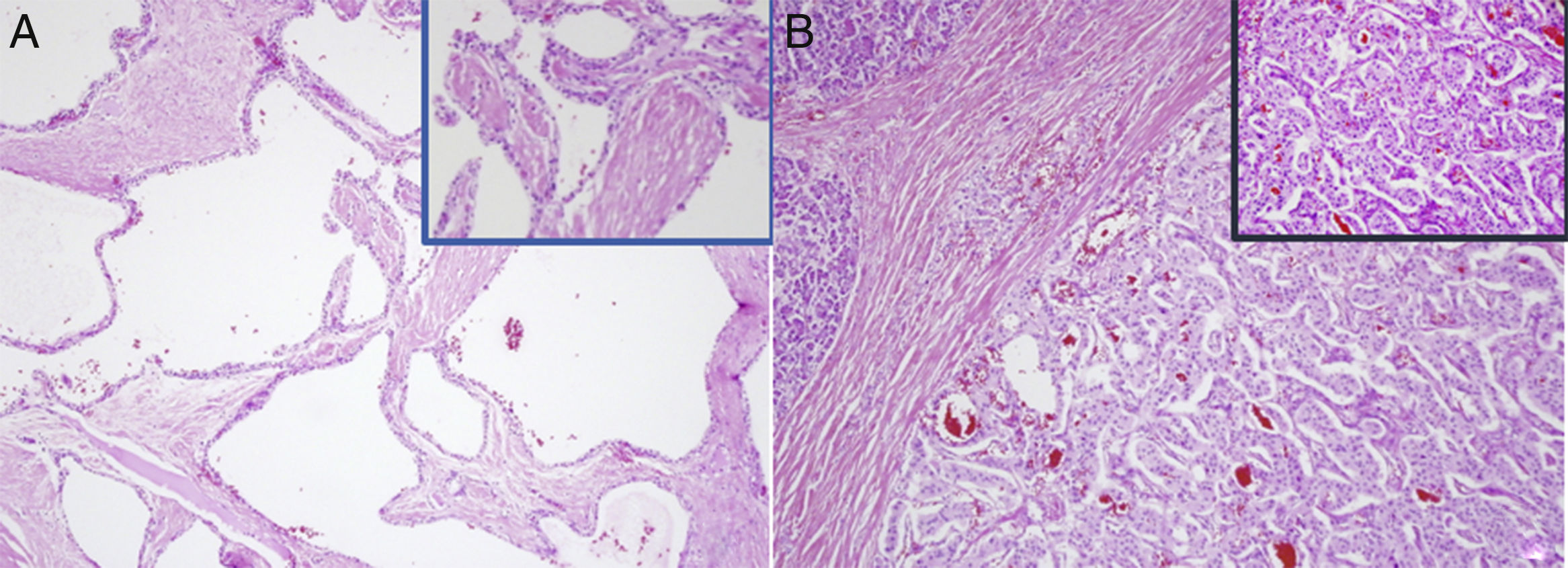
 Serous cystadenoma: multiple cystic structures, covered by cubic or cylindrical epithelial cells, with a clear cytoplasm and a round nucleus without atypia. (B) Neuroendocrine tumour: organoid proliferation of monomorphic cells, with a mildly eosinophilic cytoplasm and regular, ovoid nucleus, with fine chromatin and low mitotic activity.")
Histopathology study. (A) Serous cystadenoma: multiple cystic structures, covered by cubic or cylindrical epithelial cells, with a clear cytoplasm and a round nucleus without atypia. (B) Neuroendocrine tumour: organoid proliferation of monomorphic cells, with a mildly eosinophilic cytoplasm and regular, ovoid nucleus, with fine chromatin and low mitotic activity.
VHL disease is a rare syndrome inherited in an autosomal dominant pattern. It is caused by germ-line mutations in the VHL tumour-suppressor gene, located in the short arm of chromosome 3 (3p25). These mutations lead to the development of both benign and malignant tumours and cysts in various organs, primarily haemangioblastomas in the central nervous system (CNS) and retina, as well as renal carcinomas and pheochromocytomas.3,4 Other neoplasms have also been associated with the disease, including inner ear tumours, epididymal tumours, cystic ovarian lesions and various pancreatic tumours.3–5 The most common cause of death amongst patients with VHL disease are complications associated with CNS tumours and renal cancer, for which the life expectancy falls below 50 years of age.4
Melmon and Rosen defined the diagnostic criteria for VHL disease as the presence of a CNS haemangioblastoma and another VHL lesion, or a VHL lesion alongside a family history of VHL disease. At present, genetic testing is considered the gold standard for diagnosis.1
The risk of pheochromocytoma is used to classify VHL disease into two types, according to the kind of mutation: 1 (low risk of pheochromocytoma but high risk of other tumours), and 2 (high risk of pheochromocytoma) which is further subdivided into: 2 A: low risk for renal tumours; 2 B: high risk for renal tumours; and 2 C: pheochromocytoma alone.3,4,6 Pancreatic manifestations may be present in types 1 (without pheochromocytoma) and 2 B (with pheochromocytoma)7 – the subtype matching our case, given the patient's clinical features. Most VHL disease diagnoses are made upon the observation of haemangioblastomas during imaging tests performed as a result of neurological symptoms.
It is estimated that between 35 and 70% of VHL disease patients present with pancreatic lesions, which are often asymptomatic and detected incidentally.1,3,7,8 These are generally cystic lesions (17–56%): simple cysts or SCAs9 and, rarely, a pancreatic endocrine neoplasm (10–17%).7,9 Less frequently (11.5%), a combination of these lesions occurs, as in our case.2,3,7
In the context of VHL disease, the cystic pancreatic lesions described in the literature are benign,3,10 but can replace the pancreas and cause pancreatic insufficiency.1 The overproduction of vascular endothelial growth factor amongst these patients has been proposed as the reason behind their high prevalence.6 In general, the lesions comprise multiple cysts, distributed throughout the pancreas, and are non-functional and asymptomatic.8 Patients require a routine annual review with computed tomography (CT) or MRI to assess their progression. Surgery is only indicated if patients are experiencing symptoms and in case of complications such as infection or the compression of adjacent vessels or organs.3,5 In patients with VHL disease, SCAs typically appear in the third decade of life, while in the general population they tend to occur in women in the sixth decade.10
In VHL disease, PNETs are slow-growing, non-functional tumours and, unlike cystic lesions, may be malignant and metastatic.6 There are no clear clinical criteria for malignancy, but a size exceeding 2cm and a duplication rate of under 500 days have been proposed.4,8,10 The percentage of metastases caused by PNETs in VHL disease ranges between 13 and 36%,3,10 with the liver being the most common metastatic site.8
Monitoring in VHL disease depends on the time and type of diagnosis. In adult patients, abdominal CT scans are performed annually to monitor and detect pancreatic lesions, as well as those in the liver, adrenal glands and kidneys.2
Our patient was monitored with annual CT scans due to the presence of pancreatic cysts. VHL disease should have been suspected at this point and genetic testing been performed. However, our diagnosis was made following an intervention on a new pancreatic lesion in the head of the pancreas that had an indication for surgery, the histopathological study of which revealed a PNET and SCA. The patient therefore presented the three pancreatic lesions described in VHL disease, and her diagnosis was subsequently confirmed with genetic testing.
Cystic pancreatic lesions only require monitoring and surgery is only considered in the event of complications. However, with PNETs, surgery is proposed due to the risk of metastasis, according to tumour size.
Authors’ contributionsAylhin López Marcano: study design, acquisition and collection of data, analysis and interpretation of the results and authorship.
José M. Ramia: study design, analysis and interpretation of the results, authorship and critical review and approval of the final version.
Roberto de la Plaza: analysis and interpretation of the results, and critical review and approval of the final version.
Farah Al-Shwely, Alba Manuel Vázquez and Cristina García Amador: acquisition and collection of data and analysis and interpretation of the results.
Antonio Candia: acquisition and collection of data and analysis and interpretation of the histopathology study.
Please cite this article as: López Marcano AJ, Ramia Ángel JM, de la Plaza Llamas R, Al-Swely F, Manuel Vázquez A, García Amador C, et al. Triple afectación pancreática en una paciente con Von Hippel-Lindau. Gastroenterol Hepatol. 2018;41:446–448.
