


Craniosynostosis is a congenital anomaly resulting from the premature fusion of the cranial sutures changing growth patterns of the skull.
MethodologyFocus, scope, target population and clinical questions to be solved were defined. A systematic search for evidence in different databases (Medline, Embase, KoreaMed, Cochrane Library and the website of the World Health Organisation) in stages was performed: clinical practice guidelines; systematic reviews, and clinical trials and observational studies; using MeSH, Decs and corresponding free terms, unrestricted language or temporality. Risk of bias was evaluated using appropriate tools (AMSTAR, Risk of Bias or STROBE). The quality of evidence was graduated using the GRADE system. Modified Delphi Panel technique was used to assign the recommendation's strength and direction, as well as the degree of agreement with it, taking into account the quality of evidence, balance between risks and benefits of interventions, values and preferences of patients and availability of resources.
ResultsThere were 3712 documents obtained by search algorithms; selecting 29 documents for inclusion in the qualitative synthesis. Due to heterogeneity between studies, it was not possible to perform meta-analysis.
ConclusionsWe issued 7 recommendations and 8 good practice points, which will serve as support for decision-making in the comprehensive care of patients with craniosynostosis.
La craneosinostosis es una anomalía congénita resultante de la fusión prematura de las suturas craneales que cambia los patrones de crecimiento del cráneo.
MetodologíaSe definió el enfoque, los alcances, la población blanco y las preguntas clínicas a resolver. Se realizó una búsqueda sistematizada de la evidencia por etapas; en la primera se buscaron guías de práctica clínica, en la segunda, revisiones sistemáticas, y en la tercera, ensayos clínicos y estudios observacionales en Medline, Embase, KoreaMed, Cochrane Library y el portal de la Organización Mundial de la Salud utilizando los términos MeSH, Decs y libres correspondientes, sin restricciones de lenguaje ni temporalidad. Se evaluó el riesgo de sesgo de cada documento utilizando las herramientas AMSTAR, Risk of Bias y STROBE. Se graduó la calidad de la evidencia utilizando el sistema GRADE. Se utilizó la técnica de Panel Delphi modificada para asignar la dirección y la fuerza de la recomendación, así como el grado de acuerdo con la misma, tomando en cuenta para esto la calidad de la evidencia, el balance entre riesgos y beneficios de las intervenciones, los valores y las preferencias de los pacientes y la disponibilidad de los recursos.
ResultadosSe obtuvieron mediante los algoritmos de búsqueda 3,712 documentos, seleccionando para la inclusión en la síntesis cualitativa 29. Debido a la heterogeneidad entre los estudios no fue posible realizar un metaanálisis.
ConclusionesSe emitieron 7 recomendaciones y 8 puntos de buena práctica, los cuales servirán como apoyo para la toma de decisiones en la atención integral de pacientes con craneosinostosis.
Craniosynostosis is a common congenital anomaly, resulting from the premature fusion of the cranial sutures, which changes the growth pattern of the skull.1 It is classified as simple or compound (depending on whether one or several sutures are affected) and as primary or secondary. Primary craniosynostoses are genetic and are often present from birth. They are also divided into syndromic (familial, hereditary) and non-syndromic (isolated). Secondary craniosynostoses are due to an acquired disorder caused by a known disease, such as microcephaly, thalassaemia, sickle cell disease, metabolic or teratogenic disorders, among others. Non-syndromic cases are the most common; some can be genetic in origin, but without Mendelian inheritance. Syndromic craniosynostoses of genetic origin comprise 10%–20% of cases. More than 100 syndromes associated with craniosynostosis have been described. The overall incidence is calculated at one in 2000–2500 live births; the prevalence of all the types of craniosyntostosis, isolated and syndromic, is 343 per million. The incidence of non-syndromic craniosynostosis is approximately 0.6 per 1000 live births.2
The sagittal suture is affected in 40%–60%, the coronal suture in 20%–30% and the metopic suture in less than 10% of cases. True lambdoid synostosis is rare.3
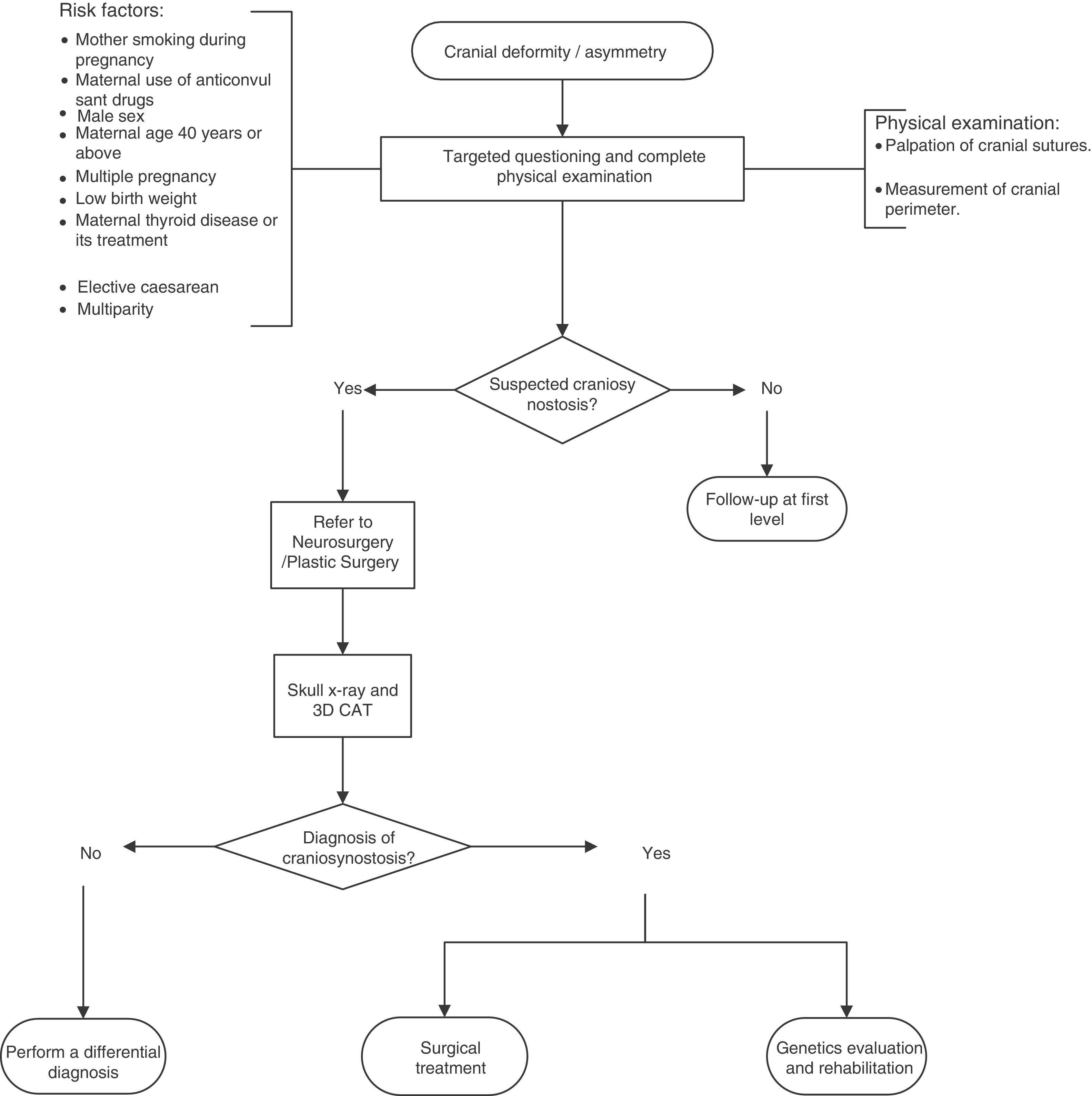
Typically, craniosynostosis is present at birth, but is not always diagnosed when it is mild. It is usually diagnosed as a cranial deformity during the first months of life. Diagnosis is based on physical examination (observing the shape of the skull and face, and on palpating the edges of the sutures and the fontanelles) and on radiological studies, including X-ray and computed tomography of the skull.2,4
It may be difficult to recognise the premature closure of the sutures; therefore it is important to monitor the cephalic perimeter to identify craniosynostosis promptly.5
Craniosynostosis can lead to 2 main groups of problems. An increase in intracranial pressure can be observed with or without hydrocephaly, although this is more common when multiple sutures are involved. Both skull and facial bone deformation can be found. For aesthetic correction or to reduce intracranial pressure, prompt corrective surgery is indicated, because 50% of skull growth is achieved by the age of 6 months.3
It is important to differentiate lambdoid synostosis from plagiocephaly (also termed occipital plagiocephaly, posterior plagiocephaly without synostosis), which is due to a postural deformation. The incidence of plagiocephaly is approximately one in 300 live births, compared to the rare incidence of lambdoid synostosis.6
Prompt assessment of a child with craniosynostosis is essential and can be started perinatally. An interdisciplinary tem is important because the correct time of intervention is critical. Moreover, coordinated care is necessary because of the complexity of medical, surgical and psychosocial factors. While early management can result in better outcomes (fewer operations and lower costs, for example), the continuity of care in a team is essential, since the results are measured through the growth and development of the child.4
There are currently no national or international clinical practice guidelines for the diagnosis, treatment and rehabilitation of patients with craniosynostosis, therefore the objective of this paper is to create the first guideline on this subject. This guideline forms part of the Master Catalogue of Clinical Practice Guidelines, with the aim of establishing a national benchmark for best available evidence-based decision making. This will improve the effectiveness, safety and quality of medical care and thus contribute to the wellbeing of people and communities – the fundamental objective and raison d’être of the health services. The Master Catalogue of Clinical Practice Guidelines can be accessed through the website of the Centro Nacional de Excelencia Tecnológica en Salud (National Centre for Health Technology Excellence) (http://www.cenetec.salud.gob.mx/contenidos/gpc/catalogoMaestroGPC.html#).
By creating and implementing this guideline we expect to achieve an increased rate of early diagnosis, establish appropriate treatment, reduce sequelae and improve the quality of life of patients with craniosynostosis.
Recommendations- •
A targeted physical examination is recommended to discount non-syndromic craniosynostosis in newborns with risk factors. Low quality of evidence/Strong recommendation.
- •
There is insufficient evidence to recommend prenatal diagnosis of craniosynostosis by ultrasound. Very low quality of evidence/No recommendation.
- •
An X-ray of the skull is recommended (anteroposterior and lateral) and computed cranial tomography with 3D reconstruction for patients with suspected craniosynostosis. Low quality of evidence/Strong recommendation.
- •
Children with bicoronal or unicoronal synostosis should undergo a molecular genetic study that includes mutations in FGFR2 exons IIIa/IIIc, FGFR3 (P250R) and TWIST1 to determine the aetiology. Low quality of evidence/Strong recommendation.
- •
Surgery should be performed on patients with craniosynostosis under the age of 12 months. Very low quality of evidence/Strong recommendation.
- •
Surgical treatment should be based on the type of craniosynostosis. Very low quality of evidence/Strong recommendation.
- •
Suturectomy and cranial reshaping or distraction with springs is recommended for patients with scaphocephaly. Very low quality of evidence/Strong recommendation.
- 1
It is recommended that an intentional search should be considered for non-syndromic craniosynostosis by means of prenatal ultrasound when there are risk factors.
- 2
The cephalic index should be determined pre- and postoperatively.
- 3
It is recommended that the parents of patients with non-syndromic craniosynostosis should be offered genetic counselling to ensure that they understand the medical aspects of the disorder, its aetiology, its probable course and possible management, as well as the risk of recurrence, for correct decision making.
- 4
The fronto-orbital advancement technique is recommended for patients with trigonocephaly and anterior plagiocephaly.
- 5
All patients with suspected non-syndromic craniosynostosis should be referred to Neurosurgery or Plastic Surgery for confirmation of diagnosis and treatment.
- 6
All patients diagnosed with non-syndromic craniosynostosis should be sent to Genetics to determine the aetiology and be offered genetic counselling.
- 7
All patients diagnosed with non-syndromic craniosynostosis should be sent to Rehabilitation for neurodevelopmental monitoring and prompt intervention to prevent neurological sequelae.
The group developing the clinical practice guideline for the diagnosis, treatment and rehabilitation of non-syndromic craniosynostosis in the 3 levels of care comprised health professionals who were specialists in physical medicine and rehabilitation, genetics, neurosurgery, general surgery and internal medicine, and physiotherapy and rehabilitation graduates, who together defined the focus, scope and clinical questions of the guideline. The target population were children aged from 0 to 23 months.
The clinical questions considered for the guideline are:
- 1
What are the risk factors for non-syndromic craniosynostosis?
- 2
What are the benchmark criteria for patients with non-syndromic craniosynostosis?
- 3
Is a prenatal diagnosis of non-syndromic craniosynostosis useful?
- 4
What is the most useful laboratory test for the diagnosis of non-syndromic craniosynostosis?
- 5
How useful are genetic studies in non-syndromic craniosynostosis?
- 6
Is genetic counselling useful for patients with non-syndromic craniosynostosis?
- 7
How useful is early surgical treatment for patients with non-syndromic craniosynostosis?
- 8
What is the surgical treatment for non-syndromic craniosynostosis?
- 9
How useful is rehabilitation management in patients with non-syndromic craniosynostosis in preventing neurological sequelae?
We undertook a systematic search of the evidence. The first stage involved a search of clinical practice guidelines in Medline, and the following websites: National Institute for Health and Care Excellence, Trip Database, Canadian Medical Association Scottish Intercollegiate Guidelines Network, Ministerio de Salud de Chile, Biblioteca del Sistema Nacional de Salud de España and the National Guideline Clearinghouse.
The second stage involved a search of systematic reviews on the following databases: Medline, Embase, and Cochrane Library. In the third stage a search was performed of primary studies specific to each question using the terms MeSH and Decs corresponding to each question in PICO format, with unrestricted language and temporality of the guide in the databases: Medline, Embase, KoreaMed, as well as the Cochrane Library and the World Health Organisation portal.
All the articles obtained through the search of the different databases algorithms were reviewed by title and abstract. The documents that referred to the subject were selected and the entire text was studied and those that met the inclusion criteria were selected for synthesis of the information.
The risk of bias of each document was evaluated using the AMSTAR7 tool for the systematic reviews, Cochrane's Risk of Bias8 tool for the clinical trial and STROBE9 for the observational studies. The quality of evidence was graded using the GRADE10 system, declaring the quality assigned together with the evidence.
A draft was prepared of the recommendations by the group developing the guide and subsequently a modified Delphi Panel11 technique was undertaken to assign the direction and strength of the recommendation, and the degree of agreement with it, taking into account the quality of evidence, the balance between the risks and benefits of the interventions, the values and preferences of the patients and the availability of the resource, obtaining over 80% approval in all of these areas. For the questions where there was insufficient evidence to make a recommendation, a point of good clinical practice was made, which was also agreed with a 100% degree of agreement.
A flow diagram was made of the diagnosis and treatment of craniosynostosis in children aged 23 months or less, based on the recommendations and points of good practice (Fig. 1).

The development group chose the most important recommendations according to their expected impact on the objectives of the guideline.
Once the final draft was completed, the content was validated by a clinical peer who was an expert in the field, and underwent interinstitutional verification by the Mexican Social Security Institute and subsequent authorisation by the National Clinical Practice Guideline Committee.
This guideline will be updated when there is evidence enough to do so, or on a scheduled basis 3–5 years after publication. The guide will be continually monitored by the users via the blog: http://cenetec-difusion.com/gpc-sns/
This guideline offers the best available evidence-based recommendations to staff of primary, secondary and tertiary care levels in order to standardise activities in relation to:
- •
Identifying the risk factors for non-syndromic craniosynostosis.
- •
Specifying the benchmark criteria for patients with non-syndromic craniosynostosis.
- •
Evaluating the usefulness of a prenatal diagnosis for non-syndromic craniosynostosis.
- •
Determining the most useful laboratory test for diagnosing non-syndromic craniosynostosis.
- •
Evaluating the usefulness of genetic studies in non-syndromic craniosynostosis.
- •
Determining the advisability of genetic counselling for patients with non-syndromic craniosynostosis.
- •
Identifying the surgical treatment for non-syndromic craniosynostosis.
- •
Establishing the usefulness of early surgical treatment of patients with non-syndromic craniosynostosis.
- •
Evaluating the usefulness of rehabilitation management in patients with non-syndromic craniosynostosis to prevent neurological sequelae.
The target users of this guideline are medical students, general nurses, medical personnel entering the social services, general doctors, resident doctors, family practitioners, paediatrics, genetics and neurosurgery specialists.
ResultsThree thousand seven hundred and twelve documents were obtained using the search algorithms, which were checked by title and abstract, reviewing the entire text of those referring to the “specific” PICO question. Twenty-nine documents were chosen for inclusion in the qualitative synthesis. Of these, 2 were systematic reviews, one a randomised clinical trial and 26 were observational studies. Five methodological documents were used as well, to substantiate the definition and justification. Due to the heterogeneity of these studies it was not possible to undertake a meta-analysis.
Recommendations and summary of evidenceRecommendation: a targeted physical examination is recommended to discount non-syndromic craniosynostosis in newborns with risk factors. Poor quality of evidence/Strong recommendation.
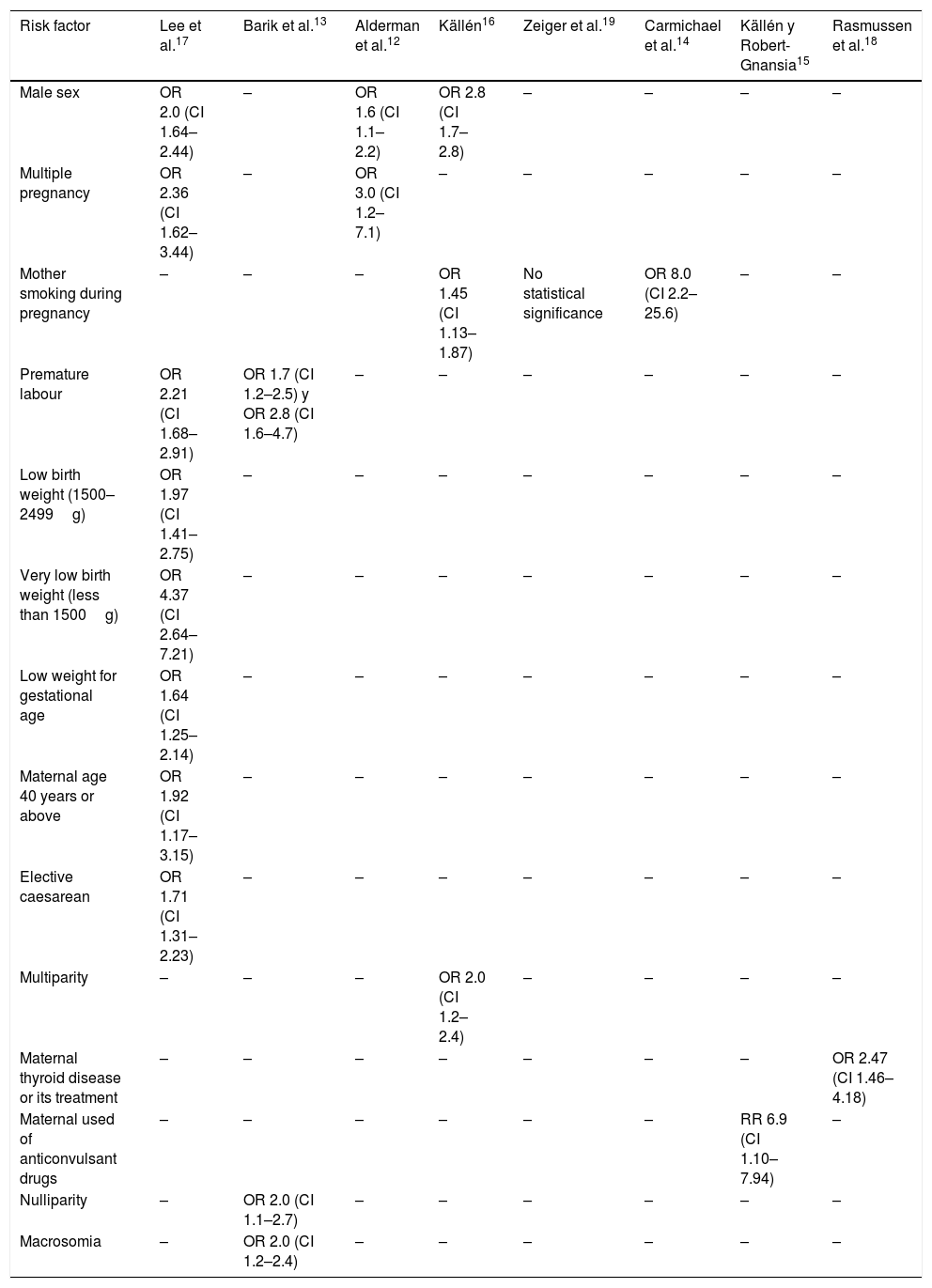
Evidence: the risk factors where a statistically significant association was found with craniosynostosis are (Table 1)12–19:
Risk factors associated with craniosynostosis.
| Risk factor | Lee et al.17 | Barik et al.13 | Alderman et al.12 | Källén16 | Zeiger et al.19 | Carmichael et al.14 | Källén y Robert-Gnansia15 | Rasmussen et al.18 |
|---|---|---|---|---|---|---|---|---|
| Male sex | OR 2.0 (CI 1.64–2.44) | – | OR 1.6 (CI 1.1–2.2) | OR 2.8 (CI 1.7–2.8) | – | – | – | – |
| Multiple pregnancy | OR 2.36 (CI 1.62–3.44) | – | OR 3.0 (CI 1.2–7.1) | – | – | – | – | – |
| Mother smoking during pregnancy | – | – | – | OR 1.45 (CI 1.13–1.87) | No statistical significance | OR 8.0 (CI 2.2–25.6) | – | – |
| Premature labour | OR 2.21 (CI 1.68–2.91) | OR 1.7 (CI 1.2–2.5) y OR 2.8 (CI 1.6–4.7) | – | – | – | – | – | – |
| Low birth weight (1500–2499g) | OR 1.97 (CI 1.41–2.75) | – | – | – | – | – | – | – |
| Very low birth weight (less than 1500g) | OR 4.37 (CI 2.64–7.21) | – | – | – | – | – | – | – |
| Low weight for gestational age | OR 1.64 (CI 1.25–2.14) | – | – | – | – | – | – | – |
| Maternal age 40 years or above | OR 1.92 (CI 1.17–3.15) | – | – | – | – | – | – | – |
| Elective caesarean | OR 1.71 (CI 1.31–2.23) | – | – | – | – | – | – | – |
| Multiparity | – | – | – | OR 2.0 (CI 1.2–2.4) | – | – | – | – |
| Maternal thyroid disease or its treatment | – | – | – | – | – | – | – | OR 2.47 (CI 1.46–4.18) |
| Maternal used of anticonvulsant drugs | – | – | – | – | – | – | RR 6.9 (CI 1.10–7.94) | – |
| Nulliparity | – | OR 2.0 (CI 1.1–2.7) | – | – | – | – | – | – |
| Macrosomia | – | OR 2.0 (CI 1.2–2.4) | – | – | – | – | – | – |
Of the newborn:
- •
Male.
- •
Very low birth weight (<1500g).
- •
Low birth weight (1500–2499g).
- •
Low weight for gestational age.
- •
Macrosomia.
Maternal:
- •
Age≥40 years.
- •
Smoking during pregnancy.
- •
Thyroid disease or treatment.
- •
Use of anticonvulsants.
- •
Multiple pregnancy.
- •
Multiparity.
- •
Nulliparity.
- •
Elective caesarean.
- •
Premature labour.
Recommendation: there is insufficient evidence to recommend prenatal diagnosis of craniosynostosis by ultrasound.20–22 Very low quality of evidence/No recommendation.
Point of good practice: it is recommended that an intentional search for non-syndromic craniosynostosis should be considered when there are risk factors.
Evidence: a case series assessed the usefulness of prenatal ultrasound for the early diagnosis of craniosynostosis in foetuses with risk factors determined by a reduction of the cranial suture space and found a sensitivity of 100% and specificity of 97% for ultrasound.20 Another case series reported a sensitivity of 58%, although the diagnosis of craniosynostosis was based on skull geometry.21 Magnetic resonance imaging was used in a case series of 15 foetuses with an abnormal prenatal ultrasound, finding 100% sensitivity and specificity when correlated with postnatal follow-up and diagnosis.22
Recommendation: a skull X-ray (anterioposterior and lateral) and computed cranial tomography with 3D reconstruction is recommended in patients with suspected craniosynostosis. Low quality of evidence/Strong recommendation.
Point of good practice: it is recommended that the cranial index should be established pre- and postoperatively.23
Evidence: the sensitivity of X-ray is 80% with a specificity of 95%, while the sensitivity of computed tomography with 3D reconstruction is 96% and specificity 100%. Bone scintigraphy is no longer used due to its low overall diagnostic precision, estimated at 66%. A diagnostic test study compared the diagnostic precision of ultrasound to that of computed tomography in 44 patients, and found that ultrasound has a sensitivity of 96.9%, specificity of 100%, positive predictive value of 100%, and negative predictive value of 92.3%.24–27
Recommendation: it is recommended that children with confirmed bicoronal or unicoronal synostosis should undergo a molecular genetic study that includes mutations in FGFR2 exons IIIa/IIIc, FGFR3 (P250R) and TWIST1 to determine the aetiology. Poor quality of evidence/Strong recommendation.
Point of good practice: it is recommended that the parents of patients with non-syndromic craniosynostosis should be offered genetic counselling to ensure that they understand the medical aspects of the disorder, its aetiology, its probable course and possible management, as well as the risk of recurrence, for correct decision making.
Evidence: a case series found that children with a clinical diagnosis of non-syndromic craniosynostosis had more genetic alterations as the cause of their synostosis if it was unicoronal or bicoronal (10 out of 48 patients) than if it was sagittal or metopic (none of the 55) (p=.0003). Moreover, it was observed that children with mutations in FGFR2 exons IIIa/IIIc, FGFR3 (P250R) and TWIST1 had more serious surgical complications than those that did not have a genetic cause (p<.05).28 Another case series included 182 patients with craniosynostosis who underwent molecular analysis of the FGFR1, FGFR2, FGFR3, TWIST1, EFNB1 and TCF12 genes, and found genetic alterations in 119 (65%).29 A systematic review showed concordance in monozygotic twins with craniosynostosis of 60.9% compared with dizygotic twins, at 5.3% (p<.0001).30
Recommendation: it is recommended that surgery should be performed on patients with craniosynostosis under 12 months of age. Very low quality of evidence/Strong recommendation.
Evidence: three studies were found that compared the intellectual quotient of patients with craniosynostosis who had undergone surgical treatment under the age of one year compared to patients operated over the age of one year, and found a statistically significant difference in IQ, which was higher in those who had been operated under the age of one: Arnaud et al. (p=.056), Bottero et al. (p=.023) and Patel et al. (p<.01).31–33 However, due to the heterogeneity of their designs it was not possible to carry out a meta-analysis. A retrospective study that included 796 patients with craniosynostosis and who had undergone surgical treatment, with the objective of determining the incidence of complications, reoperation and predictive factors, reported the following34:
Predictive factors of surgical complications:
- •
Surgical treatment performed under the age of 9 months (OR 1.88, 95% CI 1.01–3.51, p=.046).
- •
Multiple synostosis (OR 2.18, 95% CI 1.13–4.20, p=.019).
- •
Syndromic craniosynostosis (OR 2.36, 95% CI 1.31–4.25, p=.004).
- •
Younger age at the time of surgery (OR 1.05, 95% CI 1.02–1.08, p=.001).
- •
Longer duration of surgery (OR 1.44, 95% CI 1.24–1.86, p<.001).
- •
Greater amounts of blood transfusion (OR 2.97, 95%CI 1.65–5.35, p<.001).
- •
Patients who have undergone spring cranioplasty (OR 4.40, 95% CI 1.32–14.63, p=.016).
The factors associated with a greater risk of recurrent synostosis were:
- •
Syndromic craniosynostosis (p<.001).
- •
Multiple craniosynostosis (p<.001).
The factors associated with the possibility of requiring an additional surgical procedure were:
- •
Patients operated under the age of 9 months (OR 13.06, 95% CI 2.12–80.24, p<.006).
- •
Multiple synostosis (OR 11.93, 95% CI 4.07–34.94, p<.001).
- •
Syndromic craniosynostosis (OR 7.23, 95% CI 2.75–19.00, p<.001).
Recommendation: it is recommended that surgical treatment should be based on the type of craniosynostosis. Very low quality of evidence/Strong recommendation.
Evidence: in a retrospective study, the results and complications of surgery for craniosynostosis were evaluated in 283 patients treated with 12 types of surgical techniques, and the following was found35:
The most frequent postoperative complications were:
- •
Hyperthermia of unknown origin (13.43%).
- •
Infection (7.5%).
- •
Subcutaneous haematoma (5.3%).
- •
Dural tears (5%).
- •
Cerebrospinal fluid leaks (2.5%).
There were more complications in the group of reoperated patients (12.8% of the total). In the subgroup of reoperations, infection presented in 62.5%, dural tears in 93% and cerebrospinal fluid leaks in 75% of the total.
In their retrospective study that included 796 patients with craniosynostosis, Lee et al.34 reported:
- •
Total cranial vault reshaping carried a greater risk of complications (16.3%) compared to simple reshaping (6.54%) and extended reshaping (5.41%) (OR 3.4, 95% CI 1.1–10.8, p=.037).
- •
Dural fracture was associated with a greater risk of cerebrospinal fluid leak (OR 18.9, 95% CI 3.9–92.5, p<.001).
- •
Spring cranioplasty was identified as a predictor of surgical complications (OR 4.40, 95% CI 1.32–14.63, p=.016).
In their retrospective study, Kim et al. compared cranial reshaping surgery to surgery with distraction in patients with craniosynostosis, and found statistically significant differences in favour of the distraction technique in blood loss alone (p=.002), and the required volume of blood transfusion (p=.001).36
Recommendation: suturectomy and cranial reshaping or distraction with springs is recommended for patients with scaphocephaly. Very low quality of evidence/Weak recommendation.
Evidence: a systematic review that compared total cranial vault reshaping, spring cranioplasty and suturectomy for the treatment of scaphocephaly using the cephalic index, found no statistical difference for cranial reshaping compared to the spring technique, but reported a small, statistically significant difference in favour of cranial reshaping compared to open suturectomy (SMD 1.47, 95% CI 0.47–2.48). However, since there are different surgical techniques and outcomes that were not included in this review, a further review was carried out to find other studies on intervention for scaphocephaly.37
In their case series of 75 patients with scaphocephaly, David et al. compared spring-mediated cranial reshaping with cranial vault reshaping and found better outcomes in the technique with spring distraction, such as38:
- •
Less blood loss (25 compared to 255ml).
- •
Lower mean blood transfusion (0 compared to 356ml).
- •
Fewer days in the intensive care unit (0 compared to 18 days).
- •
Shorter mean hospital stay (22.7 compared to 94.2 days).
- •
Lower cost in dollars ($8339.32 compared to $27,212.49).
A retrospective study compared patients with scaphocephaly treated with spring-mediated cranioplasty with patients treated with barrel stave osteotomy; statistically significant differences were found in favour of spring-mediated cranioplasty in terms of: blood loss (p=.001), shorter hospital stay (p=.009) and shorter surgery time (p=.002). No differences in the postoperative cranial index were found between either procedure.39 A prospective study compared patients with scaphocephaly treated under the age of 6 months with endoscopic craniotomy to patients treated with total vault cranioplasty; it found that patients treated with the latter had a higher IQ (p<.05).40 The highest number of complications In scaphocephaly and synostosis of multiple sutures occurred after complete cranial vault reshaping (holocranial dismantling), followed by fronto-orbital distraction techniques.35
Recommendation: there is insufficient evidence to recommend a surgical technique for patients with trigonocephaly and anterior plagiocephaly. Very low quality of evidence/No recommendation.
Point of good practice: the fronto-orbital advancement technique is recommended for patients with trigonocephaly and anterior plagiocephaly.
Evidence: a retrospective study reported that in multiple suture synostosis, trigonocephaly and plagiocephaly, endoscopy-assisted osteotomy had a lower rate of complications, followed by fronto-orbital advancement. No other clinical trials or well-designed quasiexperimental studies were found that compared any of the surgical techniques for trigonocephaly and plagiocephaly.35
Point of good practice: all patients with suspected non-syndromic craniosynostosis should be referred to Neurosurgery or Plastic Surgery for confirmation of diagnosis and treatment.
Point of good practice: all patients with a diagnosis of non-syndromic craniosynostosis should be sent to Genetics to determine the aetiology and be offered genetic counselling.
Point of good practice: all patients with a diagnosis of non-syndromic craniosynostosis should be sent to Rehabilitation for neurodevelopmental monitoring and early intervention to prevent neurological sequelae.
Key recommendations- •
Targeted physical examination is recommended to discount non-syndromic craniosynostosis in newborns with risk factors.
- •
A skull X-ray (anteroposterior and lateral) and cranial tomography with 3D reconstruction is recommended in patients with suspected craniosynostosis.
- •
It is recommended that children with confirmed bicoronal or unicoronal synostosis should undergo a molecular genetic study that includes mutations in FGFR2 exons IIIa/IIIc, FGFR3 (P250R) and TWIST1 to determine the aetiology.
- •
It is recommended that surgery should be performed on patients with craniosynostosis under the age of 12 months.
- •
Suturectomy and cranial remodelling or spring distraction is recommended for patients with scaphocephaly.
- •
All patients with suspected non-syndromic craniosynostosis should be referred to Neurosurgery or Plastic Surgery for confirmation of diagnosis and treatment.
- •
All patients with suspected non-syndromic craniosynostosis should be sent to Genetics to determine the aetiology and be offer genetic counselling.
- •
All patients with suspected non-syndromic craniosynostosis should be sent to Rehabilitation for neurodevelopmental monitoring and early intervention to prevent neurological sequelae.
Seven recommendations and 8 points of good practice were issued in this guideline, which serve to support decision-making in the comprehensive care of patients with craniosynostosis.
NoteFor more information and to request annexes on the integration of the clinical practice guideline for the diagnosis, treatment and rehabilitation of non-syndromic craniosynostosis in the 3 care levels, visit the website of the Catálogo Maestro de Guías de Práctica Clínica, CENETEC http://www.cenetec.salud.gob.mx/contenidos/gpc/catalogoMaestroGPC.html).
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestsThis guideline was prepared with funding from the Health Secretariat of the State of Puebla, Hospital General de Puebla Eduardo Vázquez Navarro, Hospital para el Niño Poblano and Centro de Rehabilitación Infantil Teletón Puebla. The opinions of the funding entity did not influence the content of the guideline. Any conflicts of interest of members of the guideline development group have been recorded and covered by means of a declaration of the absence of any conflict of interest.
Acknowledgements go to the authorities of the Hospital General de Puebla Dr. Eduardo Vázquez Navarro, Hospital para el Niño Poblano and to the Centro de Rehabilitación Infantil Teletón Puebla for enabling the personnel of the centre or work group developing this guideline to attend the training events on evidence-based medicine and related subjects, coordinated by the Department of Health, and their support in general for the authors’ work.
Likewise, we thank the authorities of the Instituto Nacional de Pediatría (National Institute of Paediatrics) who participated in the validation and verification process, and for their valuable collaboration in preparing this guideline.
Please cite this article as: Castro Coyotl DM, Rosas Huerta XO, Sánchez Vázquez JJ, Díaz Sánchez MI, Rodríguez Peralta JS, Tetitla Munive JM, et al. Guía de práctica clínica para el diagnóstico, tratamiento y rehabilitación de craneosinostosis no sindrómica en los 3 niveles de atención. Cir Cir. 2017;85:401–410.