




La hipertensión arterial pulmonar es una complicación frecuente de las cardiopatías congénitas (CC). Es reconocido que las CC son las malformaciones más frecuentes al nacimiento con una prevalencia que va de 6 a 8 por 1,000 recién nacidos vivos. En nuestro país se calcula que cada año nacen de 12,000 a 16,000 niños con algún tipo de malformación cardiaca. En los pacientes no corregidos con cortocircuitos de izquierda a derecha el aumento de la presión pulmonar provoca incremento en las resistencias pulmonares y desencadena una disfunción endotelial y remodelación vascular, consecuencia de alteraciones en los mediadores vasoactivos que provocan vasoconstricción, inflamación, trombosis, proliferación y apoptosis celular así como fibrosis. Como consecuencia hay un incremento progresivo de las resistencias pulmonares y de la presión del ventrículo derecho. Finalmente ocurre que el flujo sanguíneo se invierte y se desarrolla el síndrome de Eisenmenger, la forma más avanzada de hipertensión arterial pulmonar consecutiva a CC. La frecuencia de hipertensión arterial pulmonar secundaria a CC ha disminuido en países desarrollados no así en países en vías de desarrollo tanto por un diagnóstico tardío como por falta de infraestructura hospitalaria o de recursos humanos para la atención de los pacientes portadores de CC. Con el advenimiento del tratamiento farmacológico para la hipertensión arterial pulmonar, se han vislumbrado nuevas oportunidades terapéuticas, siendo cada vez más cotidiano que se sumen al tratamiento intervencionista o quirúrgico en pacientes con hipertensión arterial pulmonar secundaria a CC. Se requiere conocer los factores fisiopatológicos involucrados así como llevar a cabo una cuidadosa evaluación para definir la mejor estrategia terapéutica.
Pulmonary arterial hypertension is a common complication of congenital heart disease (CHD). Congenital cardiopathies are the most frequent congenital malformations. The prevalence in our country remains unknown, based on birthrate, it is calculated that 12,000 to 16,000 infants in our country have some cardiac malformation. In patients with an uncorrected left-to-right shunt, increased pulmonary pressure leads to vascular remodeling and endothelial dysfunction secondary to an imbalance in vasoactive mediators which promotes vasoconstriction, inflammation, thrombosis, cell proliferation, impaired apotosis and fibrosis. The progressive rise in pulmonary vascular resistance and increased pressures in the right heart provocated reversal of the shunt may arise with the development of Eisenmenger’ syndrome the most advanced form de Pulmonary arterial hypertension associated with congenital heart disease. The prevalence of Pulmonary arterial hypertension associated with CHD has fallen in developed countries in recent years that is not yet achieved in developing countries therefore diagnosed late as lack of hospital infrastructure and human resources for the care of patients with CHD. With the development of targeted medical treatments for pulmonary arterial hypertension, the concept of a combined medical and interventional/surgical approach for patients with Pulmonary arterial hypertension associated with CHD is a reality. We need to know the pathophysiological factors involved as well as a careful evaluation to determine the best therapeutic strategy.
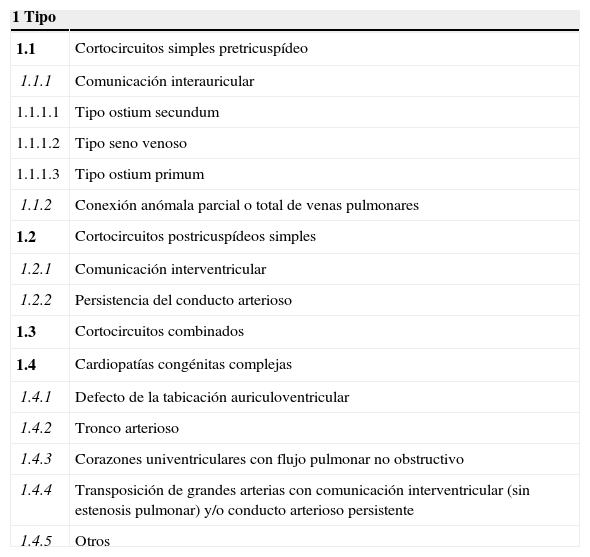
La cardiopatía congénita (CC) se define como aquella anomalía estructural evidente del corazón o de los grandes vasos intratorácicos con una repercusión real o potencial. Es bien conocido que las CC son las malformaciones más frecuentes al nacimiento con una prevalencia que va de 6 a 8 por 1,000 recién nacidos vivos1–4. Los avances en el diagnóstico y tratamiento de las CC en las pasadas décadas han dado como resultado que un importante porcentaje de pacientes con dicha dolencia alcancen la vida adulta5. La hipertensión arterial pulmonar (HAP) es una de las complicaciones más temidas en las CC; la exposición crónica de la vasculatura pulmonar a un mayor flujo como acontece en las cardiopatías con lesión pretricuspídea como la comunicación interauricular, o cortocircuitos asociados a una mayor presión como ocurre en las malformaciones postricuspídeas (p. ej. comunicación interventricular) dan como resultado la remodelación del lecho vascular lo que condiciona un incremento de las resistencias vasculares pulmonares hasta que finalmente se establece el síndrome de Eisenmenger que representa la forma más avanzada de la hipertensión pulmonar (tabla 1).
Clasificación anatomofisiopatológica de cortocircuitos sistemicopulmonares congénitos asociados a hipertensión arterial pulmonar
| 1 Tipo | |
|---|---|
| 1.1 | Cortocircuitos simples pretricuspídeo |
| 1.1.1 | Comunicación interauricular |
| 1.1.1.1 | Tipo ostium secundum |
| 1.1.1.2 | Tipo seno venoso |
| 1.1.1.3 | Tipo ostium primum |
| 1.1.2 | Conexión anómala parcial o total de venas pulmonares |
| 1.2 | Cortocircuitos postricuspídeos simples |
| 1.2.1 | Comunicación interventricular |
| 1.2.2 | Persistencia del conducto arterioso |
| 1.3 | Cortocircuitos combinados |
| 1.4 | Cardiopatías congénitas complejas |
| 1.4.1 | Defecto de la tabicación auriculoventricular |
| 1.4.2 | Tronco arterioso |
| 1.4.3 | Corazones univentriculares con flujo pulmonar no obstructivo |
| 1.4.4 | Transposición de grandes arterias con comunicación interventricular (sin estenosis pulmonar) y/o conducto arterioso persistente |
| 1.4.5 | Otros |
Modificada de Venecia 200321.
En 1897, el Dr. Víctor Eisenmenger refirió el caso de un hombre de 32 años que presentaba cianosis y disnea desde la niñez y que fallece por un episodio masivo de hemoptisis. Los hallazgos de la autopsia fueron: la presencia de un gran defecto en el tabique interventricular, hipertrofia marcada del ventrículo derecho y ateromatosis de las arterias pulmonares6. Tiempo después este síndrome fue definido y caracterizado por Paul Wood en 1958 como: «…aquella hipertensión pulmonar a nivel sistémico debida a resistencias vasculares pulmonares elevadas con un flujo bidireccional o reverso a través de un defecto septal»7. Este síndrome es un trastorno generalizado que condiciona alteraciones en la sangre, endocrinas, de la cinética de las bilirrubinas, afecta a los riñones, huesos, pulmones, lechos vasculares sistémicos y coronarios, y al sistema nervioso central.
Diversos registros europeos coinciden en señalar que son las CC de las etiologías más frecuentes que condicionan HAP. De acuerdo con la información del registro holandés de hipertensión pulmonar pediátrica recabada entre 1991-2005 y que incluyó a 3,263 pacientes, la HAP secundaria a CC representó alrededor del 80% de los casos, seguido por la HAP secundaria a problemas respiratorios8. En el registro Tracking Outcomes and Practice in Pediatric Pulmonary Hypertension (TOPP) de 317 pacientes que tenían HAP, el 36% se asoció a CC. En otro registro, el Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL), que incluyó a 216 pacientes menores de 18 años, la HAP secundaria a CC fue del 34.6% (77 pacientes)9,10.
Dependiendo de la edad de corrección de la CC, aun en aquellos pacientes en los que se logra una reparación total, existe un riesgo potencial de desarrollar HAP11. En el registro alemán, que incluye tanto a pacientes sometidos a corrección como aquellos que no lo fueron, la prevalencia de HAP varía de un 3% en pacientes con persistencia del conducto arterioso a un 100% en pacientes con ventana aortopulmonar12. El riesgo de desarrollar síndrome de Eisenmenger varía según el tipo de cardiopatía; en pacientes con comunicación interauricular no corregida es de un 10-17%, se eleva a un 50% en pacientes con comunicación interventricular y alcanza un 90% en aquellos con defecto de la tabicación auriculoventricular13. Se puede decir que los defectos con gran cortocircuito o cardiopatías complejas tienen un mayor riesgo de desarrollar en forma temprana, durante la niñez, síndrome de Eisenmenger, en comparación con los defectos como la comunicación interauricular o la conexión parcial de venas pulmonares cuya presentación es en la edad adulta.
Por lo antes señalado es evidente la importancia de una atención oportuna de los pacientes con CC. En todo el mundo se estima que anualmente nacen alrededor de 600,000 niños con una CC significativa y aproximadamente el 50% o más mueren por causa de una infección intercurrente o por insuficiencia cardiaca en la infancia y solo el 2-15% de los pacientes son llevados a intervenciones curativas. En nuestro país se llevó a cabo un censo que permitió conocer la cantidad de cirugías de CC efectuadas en 22 centros censados, entre los cuales están los más importantes; se encontró que se realizan cada año alrededor de 4,000 cirugías. Haciendo una comparación de estas cifras con otras reportadas se puede apreciar que el promedio europeo de cirugías de CC por cada millón de habitantes fue de 62. Asimismo, el promedio español de intervenciones quirúrgicas fue de 52.6. Comparativamente, el promedio mexicano de cirugías de CC por millón de habitantes fue de 38 (4,000). Este déficit ha sido parcialmente subsanado con el tratamiento por cateterismo intervencionista; sin embargo se considera que, con la información disponible, es necesario incrementar la cantidad anual de cirugías entre 1,460 y 2,540 para alcanzar los referentes español y europeo14–18.
En relación con la CC asociada con HAP CC, las guías europeas las dividen en 4 grupos (tabla 2): El primer grupo corresponde a pacientes con síndrome de Eisenmenger. En el segundo grupo se incluye a pacientes con HAP asociada a cortocircuito sistemicopulmonar, grupo en el que las cardiopatías más frecuentes son los defectos septales tanto auriculares como ventriculares. El tercer grupo lo constituyen pacientes con HAP asociada con pequeños defectos septales con un cuadro clínico y fisiopatológico similar a los pacientes con HAP idiopática. En el último grupo están aquellos pacientes que fueron llevados a cirugía pero en los que hubo progresión de la HAP a pesar de no tener defectos residuales. Está referido que este grupo de pacientes tienen un peor pronóstico con relación a los que no fueron corregidos19.
Clasificación clínica de cardiopatía congénita asociada con hipertensión pulmonar
| Síndrome de Eisenmenger |
| Hipertensión arterial pulmonar asociada a cortocircuito sistémico-pulmonar |
| Operable |
| Inoperable |
| Hipertensión arterial pulmonar asociada con pequeños defectos septales |
| Hipertensión arterial pulmonar postoperatoria |
Modificada de Galié et al.19.
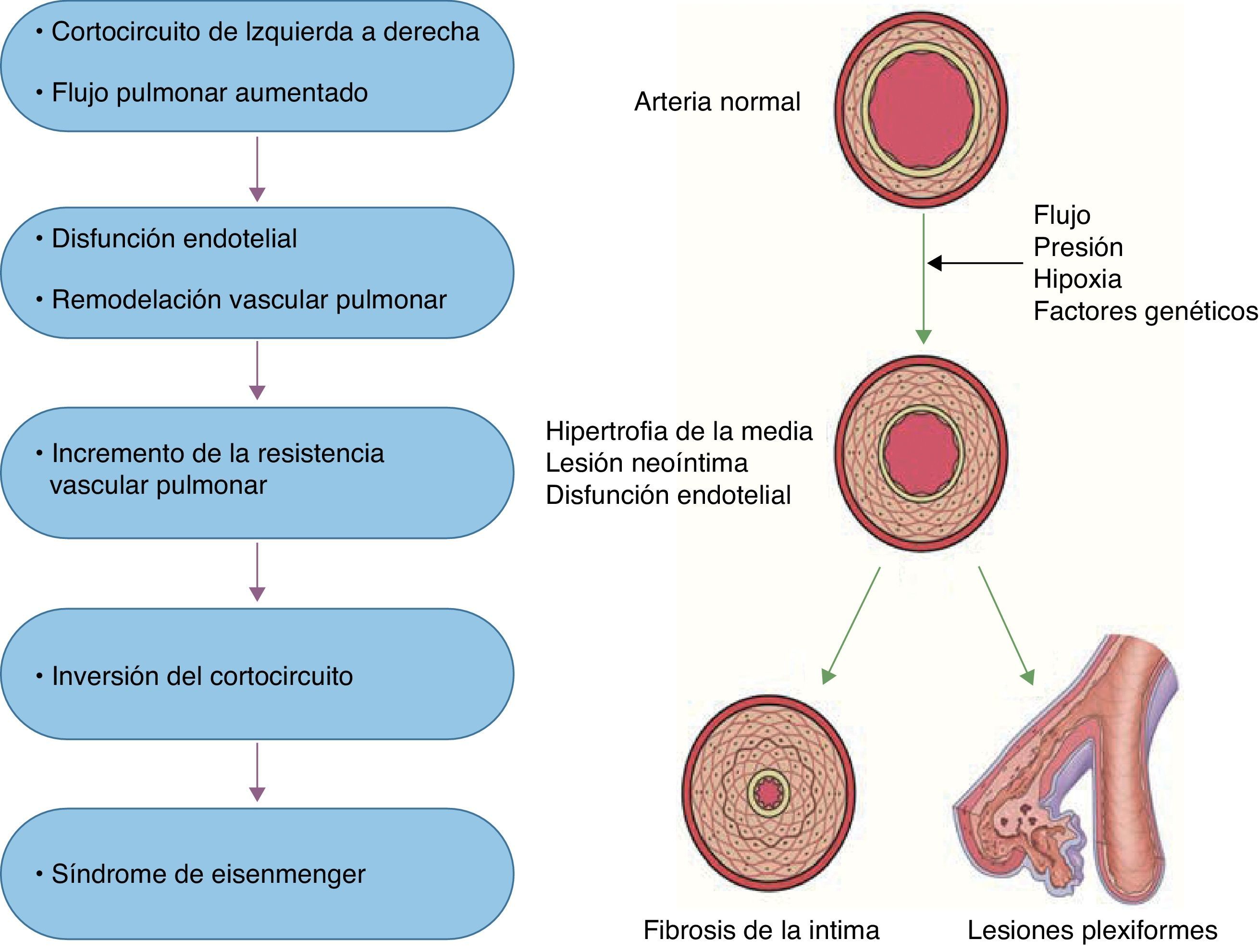
Se ha caracterizado a la HAP como resultado de diversos estímulos que condicionan disfunción endotelial, como la hipoxia sostenida, el estrés por rozamiento, la hipertensión venocapilar y otros estímulos nocivos (sustancias tóxicas, fármacos, inflamación), que condicionan cambios vasculares estructurales que determinan una remodelación anormal de la circulación pulmonar principalmente a nivel de las arteriolas precapilares20 (fig. 1). En esta remodelación patológica participan la célula endotelial, la del músculo liso vascular y los elementos que constituyen la matriz extracelular en la adventicia. Es mediada por múltiples sustancias con potencial efecto vasoconstrictor y/o de proliferación o por la falta de acción de mediadores vasodilatadores, antiproliferativos o ambos. Su desequilibrio determina lesiones vasculares: hipertrofia de la media muscular del vaso, extensión de músculo hacia la periferia del vaso (neomuscularización), proliferación (celular o fibromuscular) de la íntima y trombosis in situ. En la trombosis in situ participan tanto un aumento de factores protrombóticos (tromboxano A2, endotelina) como una disminución de factores antiagregantes (prostaciclina, óxido nítrico y trombomodulina). La vasoconstricción, la trombosis in situ y la remodelación vascular anormal descrita condicionan la obstrucción de los vasos pulmonares a nivel microvascular y explican el incremento de la resistencia vascular y de la presión arterial pulmonar. Como consecuencia de lo anterior, el incremento sostenido de la poscarga ventricular derecha, de manera aislada o en asociación con otros factores, conduce a la falla ventricular derecha y eventualmente a la muerte21.

Las alteraciones histopatológicas características fueron descritas por Heath y Edwards22 en 1958 por medio de una clasificación que establece 6 grados (i-vi) de vasculopatía pulmonar. Cabe hacer mención que la clasificación no implica necesariamente un carácter evolutivo. Sin embargo, la severidad del compromiso hemodinámico (elevación de la resistencia vascular pulmonar a nivel suprasistémico) suele aparecer en presencia de lesión vascular de grado iii (infiltración fibromuscular de la íntima) en adelante. En 1978, la Dra. Marlene Rabinovitch describió una modificación a esta clasificación en especímenes de biopsia de pulmón mediante un método cuantitativo del análisis morfométrico de la enfermedad vascular en pacientes con CC con correlación hemodinámica. Esta clasificación comprendía 3 estadios evolutivos (A, B y C) caracterizados por: hipertrofia de la media, neomuscularización arterial (extensión anormal de la capa muscular) y, por último, una cantidad reducida y diámetro de las arterias23,24.
Respecto a la remodelación vascular, se ha propuesto que la presión y el flujo elevado condicionen daño al endotelio pulmonar lo que a su vez altere la permeabilidad de la lámina elástica. Ello puede estar asociado a la degradación de matriz extracelular por activación de la enzima elastasa endovascular y metaloproteinasas de matriz, que junto a la liberación del factor transformador de crecimiento beta (TGF-β) y factor de crecimiento de fibroblastos activan la tenascina, una glucoproteína que amplifica la señal para la hipertrofia/proliferación de células de músculo liso vascular y fibroblastos. Finalmente, las células de músculo liso vascular modifican su fenotipo contráctil a migratorio en presencia de otra glucoproteína, la fibronectina, con lo que migran y proliferan en la luz del vaso. En un modelo experimental con HAP se pudo demostrar que cuando la elastasa endovascular es inhibida, declinan las concentraciones de tenascina y ocurre apoptosis (muerte celular programada) con lo que la vasculopatía pulmonar involuciona25.
Las proteínas morfogenéticas óseas (BMP) son miembros de la superfamilia del TGF-β. Son cinasas multifuncionales que regulan el crecimiento celular, la proliferación y la apoptosis. Existe evidencia creciente de que la disminución en la expresión o disfunción del receptor 2 de la BMP (BMPR2) participa en el desarrollo de HAP. Esto implica que un trastorno en el proceso de señalización del crecimiento celular reduce la función de freno de BMPR2 (antiproliferativa), y de manera simultánea causa la activación de otros receptores de TGF-β que estimulan la proliferación de músculo liso en las arteriolas pulmonares. Los receptores de BMPR2 localizados en la membrana celular son cinasas de serina que actúan en secuencia y que al ser fosforilados (activados) envían señales mediante las proteínas Smad, lo cual permite la traslocación del núcleo, unirse al ADN y regular la transcripción genética. El desequilibrio BMP/TGF-β provocaría efectos proliferativos y antiapoptósicos26,27.
La susceptibilidad genética se ha vinculado con el desarrollo de HAP y enfermedad vascular obstructiva crónica. Roberts et al.28 identificaron la existencia de mutaciones en el gen del BMPR2, miembro de la superfamilia TGF-β, en un 6% de los pacientes con HAP asociada a CC. Cabe señalar que dicha mutación no ha sido investigada en niños y adultos con CC sin HAP por lo que es imposible actualmente diferenciar el papel que desempeña el hiperflujo y la presión pulmonar en comparación con la mutación genética en la aparición de la enfermedad vascular obstructiva pulmonar. Como extrapolación a la presencia de mutaciones al gen BMPR2 como responsable en la HAP idiopática y hereditaria en donde ciertamente es mucho más frecuente (26% y 70% respectivamente), la identificación de mutaciones al gen BMPR2 puede ser solo un factor de riesgo en el caso de HAP asociada a CC y depende de la presencia de otros «modificadores» genéticos o ambientales para desarrollar HAP clínica. Es evidente que hacen falta más estudios para determinar su verdadero papel.
El hiperflujo pulmonar secundario al cortocircuito de izquierda a derecha aumenta las fuerzas de rozamiento y el estrés circunferencial de la pared vascular e induce disfunción endotelial. Celermajer et al.29 en 1993 describieron trastornos de relajación-derivado del endotelio de la arteria pulmonar en niños con CC e hiperflujo pulmonar. Por otra parte, en pacientes con síndrome de Eisenmenger se han observado concentraciones elevadas tisular y plasmática de endotelina 130–32. Pero la vasoconstricción sostenida también es promovida por disfunción de los canales de potasio. Estos son proteínas transmembrana que poseen gran selectividad para el potasio y existen varios tipos. Los que tienen mayor relevancia son los canales Kv que se activan por voltaje y contribuyen al potencial transmembrana de la célula de músculo liso vascular. Al ser inhibidos, se acumula potasio dentro de la célula lo que provoca su despolarización. Al cambiar el potencial (se vuelve más positivo), se abren los canales tipo L para el calcio y aumenta la concentración de calcio libre en el citosol que activa el aparato contráctil, promoviendo vasoconstricción y proliferación celular33–36. Machado et al.36 informaron de la inhibición de los Kv en cultivo de células de músculo liso vascular de sujetos normales al adicionar suero de pacientes con HAP asociada a CC33–37.
Además, como resultado del daño que sufre el endotelio pulmonar existe inflamación, lo cual favorece la adherencia y activación de plaquetas y leucocitos, y por ende, la presentación de fenómenos protrombóticos que contribuye a la aparición de obstrucción vascular pulmonar.
Proceso diagnósticoLas pruebas que deben realizarse en estos pacientes se deciden por las manifestaciones clínicas (tabla 3) y los recursos disponibles (tabla 4). Los objetivos de estos estudios incluyen: 1) Confirmar el diagnóstico de HAP y su gravedad, 2) Caracterizar correctamente las lesiones cardiacas subyacentes, 3) Excluir cualquier potencial causa añadida de elevación de resistencia vascular pulmonar e hipoxia, 4) evaluar la extensión de la disfunción multiorgánica y 5) establecer de manera objetiva el estatus funcional basal38.
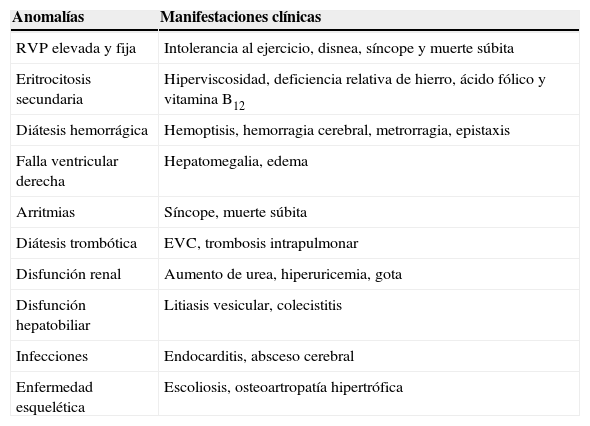
Anomalías frecuentes en el síndrome de Eisenmenger y su traducción clínica
| Anomalías | Manifestaciones clínicas |
|---|---|
| RVP elevada y fija | Intolerancia al ejercicio, disnea, síncope y muerte súbita |
| Eritrocitosis secundaria | Hiperviscosidad, deficiencia relativa de hierro, ácido fólico y vitamina B12 |
| Diátesis hemorrágica | Hemoptisis, hemorragia cerebral, metrorragia, epistaxis |
| Falla ventricular derecha | Hepatomegalia, edema |
| Arritmias | Síncope, muerte súbita |
| Diátesis trombótica | EVC, trombosis intrapulmonar |
| Disfunción renal | Aumento de urea, hiperuricemia, gota |
| Disfunción hepatobiliar | Litiasis vesicular, colecistitis |
| Infecciones | Endocarditis, absceso cerebral |
| Enfermedad esquelética | Escoliosis, osteoartropatía hipertrófica |
EVC: evento vascular cerebral; RVP: resistencia vascular pulmonar.
Modificada de Krishna-Kumar y Sandoval34.
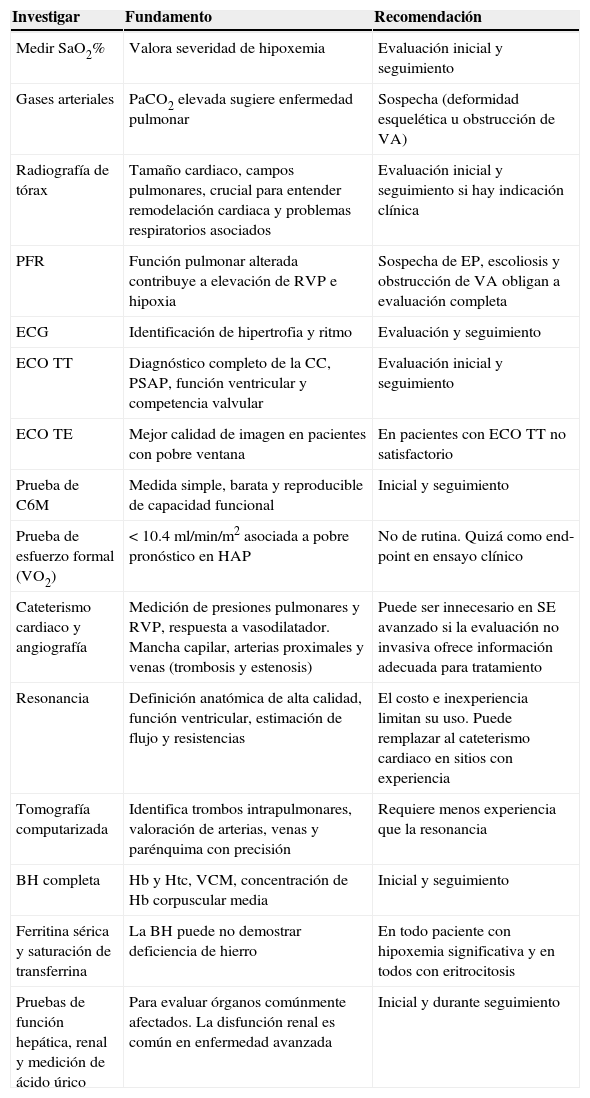
Abordaje diagnóstico propuesto
| Investigar | Fundamento | Recomendación |
|---|---|---|
| Medir SaO2% | Valora severidad de hipoxemia | Evaluación inicial y seguimiento |
| Gases arteriales | PaCO2 elevada sugiere enfermedad pulmonar | Sospecha (deformidad esquelética u obstrucción de VA) |
| Radiografía de tórax | Tamaño cardiaco, campos pulmonares, crucial para entender remodelación cardiaca y problemas respiratorios asociados | Evaluación inicial y seguimiento si hay indicación clínica |
| PFR | Función pulmonar alterada contribuye a elevación de RVP e hipoxia | Sospecha de EP, escoliosis y obstrucción de VA obligan a evaluación completa |
| ECG | Identificación de hipertrofia y ritmo | Evaluación y seguimiento |
| ECO TT | Diagnóstico completo de la CC, PSAP, función ventricular y competencia valvular | Evaluación inicial y seguimiento |
| ECO TE | Mejor calidad de imagen en pacientes con pobre ventana | En pacientes con ECO TT no satisfactorio |
| Prueba de C6M | Medida simple, barata y reproducible de capacidad funcional | Inicial y seguimiento |
| Prueba de esfuerzo formal (VO2) | <10.4ml/min/m2 asociada a pobre pronóstico en HAP | No de rutina. Quizá como end-point en ensayo clínico |
| Cateterismo cardiaco y angiografía | Medición de presiones pulmonares y RVP, respuesta a vasodilatador. Mancha capilar, arterias proximales y venas (trombosis y estenosis) | Puede ser innecesario en SE avanzado si la evaluación no invasiva ofrece información adecuada para tratamiento |
| Resonancia | Definición anatómica de alta calidad, función ventricular, estimación de flujo y resistencias | El costo e inexperiencia limitan su uso. Puede remplazar al cateterismo cardiaco en sitios con experiencia |
| Tomografía computarizada | Identifica trombos intrapulmonares, valoración de arterias, venas y parénquima con precisión | Requiere menos experiencia que la resonancia |
| BH completa | Hb y Htc, VCM, concentración de Hb corpuscular media | Inicial y seguimiento |
| Ferritina sérica y saturación de transferrina | La BH puede no demostrar deficiencia de hierro | En todo paciente con hipoxemia significativa y en todos con eritrocitosis |
| Pruebas de función hepática, renal y medición de ácido úrico | Para evaluar órganos comúnmente afectados. La disfunción renal es común en enfermedad avanzada | Inicial y durante seguimiento |
BH: biometría hemática; CC: cardiopatía congénita; C6M: caminata de 6min; ECO TE: ecocardiograma transesofágico; ECO TT: ecocardiograma transtorácico; ECG: electrocardiograma; EP: enfermedad pulmonar; HAP: hipertensión arterial pulmonar; Hb: hemoglobina; Htc: hematocrito; PaCO2: presión de dióxido de carbono; PFR: pruebas de función respiratoria; PSAP: presión sistólica de arteria pulmonar; RVP: resistencias vasculares pulmonares; SaO2: saturación arterial de oxígeno; SE: síndrome de Eisenmenger; VA: vía aérea; VCM: volumen corpuscular medio; VO2: consumo de oxígeno.
Modificada de Krishna-Kumar y Sandoval38.
La posibilidad de que los pacientes con CC e hipertensión pulmonar puedan ser tratados por cateterismo intervencionista o cirugía tiene que ser perfectamente determinada desde el punto de vista clínico y apoyada en estudios de imagen y que evalúan de manera dinámica la vasorreactividad del lecho pulmonar. Cuando se palpa el cierre de la pulmonar y hay presencia de chasquido de apertura pulmonar, ha desaparecido el soplo previamente existente, hay cianosis manifiesta, dedos hipocráticos, edema periférico y ascitis, es evidente que el paciente desarrolló un síndrome de Eisenmenger y no es candidato a manejo quirúrgico convencional.
Otro grupo lo constituyen los que en la evaluación clínica y en un estudio ecocardiográfico se pueda definir sin lugar a dudas su operabilidad. También existe un tercer grupo que se ubica en una zona gris y es en el que hay un gran problema para llegar a una decisión terapéutica y donde se deben tener todas las herramientas disponibles para definir la mejor conducta, ya sea su eventual tratamiento quirúrgico o el mantener los pacientes en seguimiento clínico.
El estudio ecocardiográfico es el principal estudio no invasivo para definir el diagnóstico del tipo de la CC y a la vez permite la evaluación de aspectos funcionales y hemodinámicos, y tienen una buena correlación con los estudios de cateterismo cardiaco pero no siempre es suficiente para determinar la operabilidad en los pacientes limítrofes. Es en estos pacientes en los que el cateterismo cardiaco es el estudio fundamental para determinar los aspectos hemodinámicos y de vasorreactividad.
Definir la operabilidad de un niño con CC relacionada con hipertensión pulmonar es conjuntar información que nos permita predecir su evolución favorable o desfavorable. Dentro de las herramientas que han sido utilizadas con ese fin está la biopsia pulmonar para hacer el análisis histopatológico. Sin embargo, ha dejado de ser un parámetro para definir la operabilidad tanto por el riesgo inherente al procedimiento como por el hecho de que el área donde se toma la muestra del parénquima pulmonar no es representativa de las alteraciones en ambos pulmones. Algunos pacientes desarrollan enfermedad vascular pulmonar avanzada a pesar de que la biopsia de tejido pulmonar mostraba lesiones moderadas o poco significativas, mientras que otros pacientes con lesiones graves determinadas por el estudio histopatológico, una vez resuelta la cardiopatía quirúrgicamente tienen una evolución adecuada a medo y largo plazo lo que apoya la reversibilidad de las lesiones39.
La realización del cateterismo derecho para la evaluación hemodinámica así como de pruebas de reto para determinar la presencia de vasorreactividad, siguen siendo una de las principales herramientas para definir la operabilidad y la posible evolución postoperatoria a corto y largo plazo. El estudio de la respuesta vascular se inicia en la década de los 70 del siglo pasado. En 1978 se describieron los efectos de la administración de isoproterenol sobre la reactividad vascular pulmonar, después de la nifedipina, del diazóxido y de la hidralazina40–45. En épocas recientes se agregaron a esta lista la adenosina, el óxido nítrico y el iloprost. La presencia de resistencias vasculares pulmonares≤6UW/m2 o una relación de resistencias sistémicas-pulmonares≤0.3 son predictores de una buena evolución postoperatoria. Las pruebas de reto farmacológico para definir la presencia de vasorreactividad se realizan cuando las resistencias vasculares pulmonares están en 6-9UW/m2 o la relación de resistencias se encuentra entre 0.3-0.5 (tabla 5)46,47. Es necesario puntualizar la existencia de diversos aspectos técnicos en la medición de las variables hemodinámicas en pacientes con CC con cortocircuitos aislados, por ejemplo una comunicación interventricular, que pueden provocar sesgo en la información obtenida. Esto se vuelve más evidente en pacientes con cardiopatías complejas, como el tronco común arterioso, en donde los cálculos revisten una mayor incertidumbre dado que algunos parámetros se basan en valores teóricos y no medidos; por ejemplo, el consumo de oxígeno, lo que hace que la decisión de la conducta terapéutica sea muy difícil. Algunos autores han considerado que no existen parámetros hemodinámicos que nos puedan definir, con toda certeza, el grado de riesgo, llámese muerte o resistencias vasculares pulmonares elevadas persistentes, después de una corrección quirúrgica, y que ello depende de múltiples factores, desde el tipo de malformación cardiaca hasta de predisposición genética48.
Criterios para definir la operabilidad de pacientes con HAP secundaria a cardiopatía congénita
| Aspectos hemodinámicos | |
|---|---|
| Basal | RVP≤6UW/m2RVP/RVS≤0.3 |
| Indicación reto farmacológico O2, ON, adenosina, iloprost | BasalRVP≥6-9UW/m2RVP/RVS0.3-0.5 |
| Reactividad vascular | Disminución de RVP o de la PAM del 20%Disminución del 20% de índice RVP/RVSRVP≤6UW/m2RVP/RVS≤0.3 |
HAP: hipertensión arterial pulmonar; ON: óxido nítrico; PAPm: presión pulmonar media; RVP: resistencias vasculares pulmonares; RVS: resistencias vasculares sistémicas.
Modificada de Myers et al.47
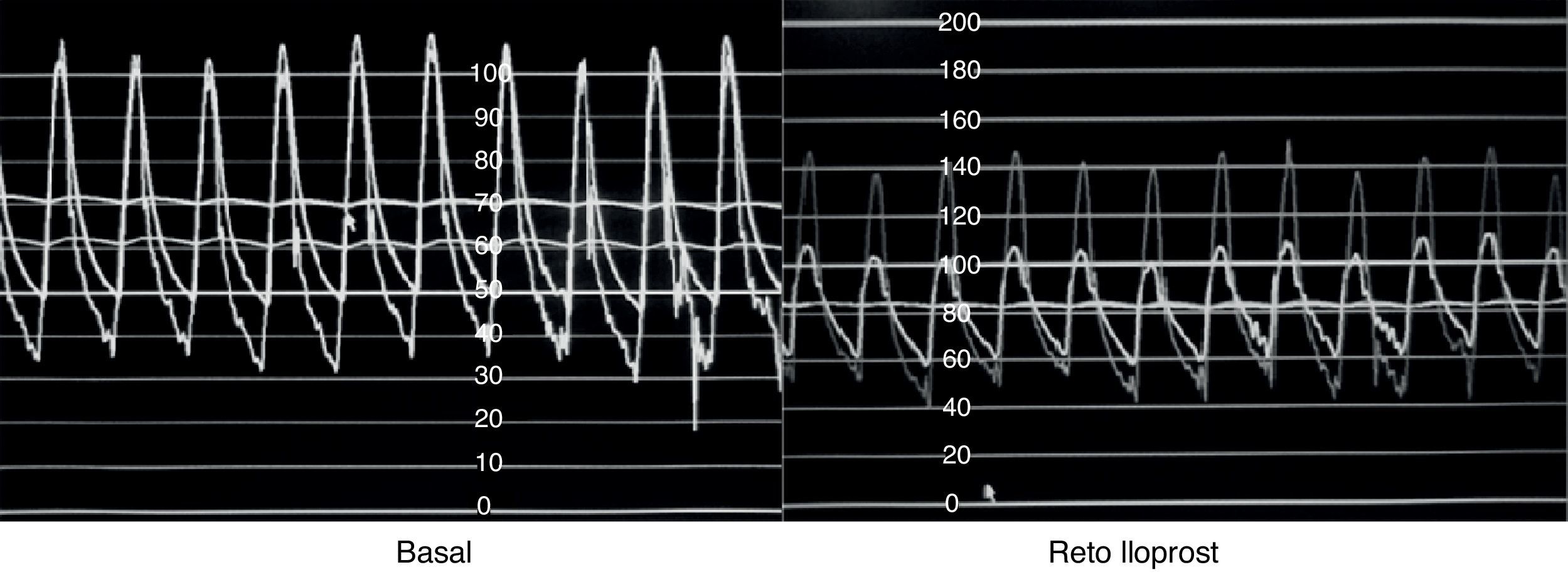
Se han utilizado diversos manejos farmacológicos durante la realización del cateterismo cardiaco cuyo objetivo es definir la presencia de vasorreactividad pulmonar, destacando el oxígeno al 100%, el isoproterenol, la adenosina, y en épocas recientes, el óxido nítrico y el iloprost49,50. Se habla de que la asociación de oxígeno y óxido nítrico confiere una sensibilidad del 97% y una especificidad del 90% para predecir la operabilidad tomando como punto de corte una relación sistemicopulmonar≤0.3, en contraposición de cuando el manejo es solo con oxígeno en el que la sensibilidad y la especificidad son del 64% y del 68%, respectivamente. En cuanto al prostanoide iloprost, diversas investigaciones han demostrado un efecto vasodilatador importante similar al que producen el óxido nítrico, la adenosina y las prostaciclinas lo que podría abrir la posibilidad de su uso para identificar a los pacientes con respuesta a estos fármacos (fig. 2). Sin embargo, aún carecemos de evidencia suficiente que valide su utilización como fármaco para determinar la vasorreactividad pulmonar pero consideramos que es factible la realización de un estudio multicéntrico, en nuestro medio, para explorar que se adopte como la prueba idónea para definir la operabilidad de los pacientes con HAP secundaria a CC51–54.
.")
Dentro de la evaluación de todo niño con hipertensión pulmonar significativa secundaria a CC estamos convencidos de que se debe realizar angiografía pulmonar en cuña amplificada para definir la gravedad de la HAP y la operabilidad del paciente. Los antecedentes de esta herramienta comienzan en 1958 cuando Evans y Short llevan a cabo una angiografía pulmonar magnificada post mórtem, y 20 años después Nihill y McNamara aplican los avances tecnológicos en radiología y describen una técnica sencilla, y finalmente en 1981, Rabinovitch et al.55, con una técnica similar, realizan un análisis cuantitativo con correlación hemodinámica e histológica. La evaluación que se lleva a cabo con el estudio es la relación del tamaño de la arteria y vena centrales, la tortuosidad de los vasos pulmonares y la disminución del calibre del vaso de 2.5 a 1.5mm. Cuando esto ocurre en una longitud≥13mm es normal, cuando es<10mm se habla de una hipertrofia severa de la media, y <7mm demuestra además hiperplasia de la íntima; cantidad de arterias monopediales, las cuales son vasos distales, generalmente terminales, que nacen en un ángulo de 90° de la arteria central y miden <700μ. Al haber hiperplasia y fibrosis de la íntima disminuye el calibre de estos vasos y termina por ocluirlos disminuyendo la cantidad; con relación a la mancha capilar que se relaciona con la cantidad de arterias intraacinares, la oclusión de estas arterias se produce al azar por lo que la mancha capilar tendrá un patrón heterogéneo en la enfermedad oclusiva avanzada, la mancha capilar desaparece y se podrán observar zonas avasculares; el tiempo de circulación que transcurre entre el inicio de la inyección y la aparición del contraste en la vena central principal o la aurícula izquierda es mayor a medida que aumenta la resistencia vascular pulmonar, reflejo de grados mayores de lesión vascular. El estudio se realiza con un catéter con orificio distal en una arteria de la periferia pulmonar (lóbulo inferior derecho e izquierdo) en posición semienclavada y se puede llevar a cabo correlación con la clasificación de Heath y Edwards22. En pacientes sin lesión vascular pulmonar, las arterias musculares son normales con una relación veno-arterial>1, existe una disminución gradual del calibre (>13mm), las arterias monopediales son numerosas y la mancha capilar homogénea, y la rapidez del tiempo de circulación ≤0.5 seg; y en contraposición, la presencia de arterias musculares dilatadas con una relación veno-arterial<1 y tortuosidad generalizada con disminución abrupta del calibre (<7mm), arterias monopediales casi ausentes y una mancha capilar irregular y tenue (grandes zonas avasculares) colocaría al paciente en una clasificación de Heath y Edwards IV (fig. 3).
.")
Angiografía pulmonar en cuña magnificada donde se puede observar diversas manchas capilares. En el extremo izquierdo hay una mancha capilar normal y en el extremo derecho se aprecia el llamado árbol de invierno, de un paciente con HAP grave con resistencias vasculares fijas. Las imágenes intermedias muestran diversos grados de afectación vascular. (Imágenes cortesía de los Dres. José A. García-Montes y Cecilia Britton).
Se tiene como premisa que en todo paciente con síndrome de Eisenmenger no existe ningún grado de reactividad vascular pulmonar, es decir sus resistencias son fijas. Sin embargo, existe información consistente en la cual se demuestra que alrededor de un tercio de los pacientes pueden mantener algún grado de vasorreactividad pulmonar lo que queda manifiesto al ser tratados con óxido nítrico a dosis de 20-80ppm sin cambios en la presión sistémica y con incremento en el GMPc56. La importancia de este hallazgo aumenta con el estudio de Post et al. en el que se encontró que los pacientes con síndrome de Eisenmenger que tenían vasorreactividad tenían una mayor supervivencia57.
En muchos campos de la medicina el uso de biomarcadores ha sido un elemento que ha permitido predecir desenlaces y la hipertensión pulmonar no ha sido ajena a ello. Se han utilizado diversos biomarcadores como ácido úrico, péptido natriurético, troponina cardiaca T, metabolitos urinarios de prostaglandinas, relación de endotelina 1 y 3, factor de von Willebrand, células endoteliales circulantes (cec) y micro-ARN, entre otros58–62. Las cec son un nuevo biomarcador prometedor que indica daño y remodelación vascular. En el estudio inicial, que incluyó biopsia pulmonar, Smadja et al.61 mostraron que el número de cec era significativamente mayor en pacientes con CC e HAP irreversible (57cec/ml) que en aquellos con CC e HAP reversible o en los controles sin HAP (3cec/ml). EN función de estos hallazgos, que indicarían daño endotelial sostenido en los pacientes con CC e HAP irreversible, uno podría predecir la respuesta hemodinámica posterior al cierre del defecto. En un estudio posterior, el mismo grupo de investigadores demostró la utilidad de este biomarcador en el pronóstico y en el seguimiento (respuesta al tratamiento médico) en pacientes con CC e HAP irreversible al igual que en pacientes con HAP idiopática. De confirmarse estos hallazgos, dispondríamos de un biomarcador muy útil para el tratamiento de estos pacientes. Asimismo, las micro-ARN son pequeñas moléculas que regulan la expresión genética a nivel de la postranscripción; si bien hay estudios en los que niveles de micro-ARN se han relacionado con mortalidad a 2 años, no existen aún investigaciones que muestren que este biomarcador puede diferenciar entre reversibilidad e irreversibilidad de la HAP en pacientes con CC63.
TratamientoEl tratamiento de hipertensión pulmonar asociada a CC y síndrome de Eisenmenger se basa en experiencias clínicas más que en estudios ex profeso diseñados para lograr un nivel de evidencia sólido (tabla 6). Hasta hace poco tiempo era muy limitado lo que se podía ofrecer a este tipo de pacientes64. En estos pacientes, con resistencia vascular pulmonar elevada, la resistencia sistémica debe ser mayor para permitir un adecuado flujo pulmonar a pesar de un cortocircuito de derecha a izquierda. Si la resistencia vascular sistémica disminuye de manera súbita el cortocircuito venoarterial aumenta y puede condicionar una hipoxemia fatal. Las medidas generales que se indican van dirigidas a evitar diversas situaciones clínicas como es el embarazo, la deshidratación, exposición prolongada al calor, fiebre prolongada, buceo, el ejercicio intenso así como la exposición crónica a grandes alturas y la deficiencia de hierro.
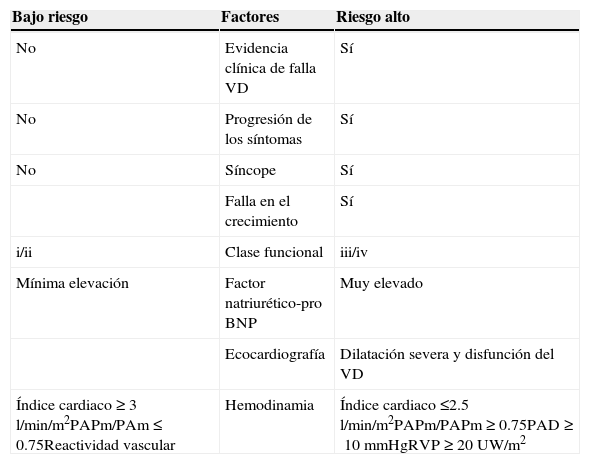
Factores de riesgo en paciente pediátrico
| Bajo riesgo | Factores | Riesgo alto |
|---|---|---|
| No | Evidencia clínica de falla VD | Sí |
| No | Progresión de los síntomas | Sí |
| No | Síncope | Sí |
| Falla en el crecimiento | Sí | |
| i/ii | Clase funcional | iii/iv |
| Mínima elevación | Factor natriurético-pro BNP | Muy elevado |
| Ecocardiografía | Dilatación severa y disfunción del VD | |
| Índice cardiaco≥3l/min/m2PAPm/PAm≤0.75Reactividad vascular | Hemodinamia | Índice cardiaco≤2.5l/min/m2PAPm/PAPm≥0.75PAD≥10mmHgRVP≥20UW/m2 |
BNP: péptido natriurético tipo B; PAD: presión aurícula derecha; PAm: presión sistémica media; PAPm: presión pulmonar media; RVP: resistencias vasculares pulmonares; VD: ventrículo derecho.
Modificada de Ivy et al.64.
No se ha demostrado que el oxígeno suplementario modifique la supervivencia de los pacientes con síndrome de Eisenmenger, al menos cuando se suministra durante la noche, por lo que su uso solo se justifica en aquellos pacientes que tienen un efecto evidente al incrementar la saturación de oxigeno (del 5 al 10%) y observar mejoría clínica65.
La eritrocitosis secundaria es una respuesta fisiológica mediada por la eritropoyetina como respuesta a la hipoxemia crónica. La flebotomía no debe considerarse un procedimiento de rutina en niños con síndrome de Eisenmenger. Solo está indicada en pacientes con síntomas de hiperviscosidad significativos, que casi siempre se observan con hematocritos de≥70%, como: cefalea, tinnitus, parestesias, trastornos visuales, mialgias, etc. En aquellos pacientes sintomáticos en quienes esté indicada su realización, no es adecuado extraer grandes volúmenes sanguíneos pues tendrán que ser restituidos con solución salina por los menos con un volumen similar. En pacientes adolescentes y adultos se sugiere una cantidad entre 300-500ml de sangre, y en niños en edad preescolar y escolar de acuerdo con su peso y talla. Es recomendable la utilización de filtros de aire para prevenir el riesgo de embolismo. La realización repetida de flebotomías provoca una mayor actividad medular y depleción de los depósitos de hierro lo cual condiciona, finalmente, un círculo vicioso al producir mayor hiperviscosidad. Es conveniente recordar que la eritrocitosis secundaria no es un factor de riesgo para accidente vascular cerebral, por lo que la concentración de hemoglobina no debe ser por sí misma el indicador para la realización de flebotomías. En todo paciente con síntomas de hiperviscosidad se debe estar seguro de que no esté deshidratado, tenga anemia relativa por déficit de hierro o la sintomatología sea secundaria a un absceso cerebral66–69 (tabla 7).
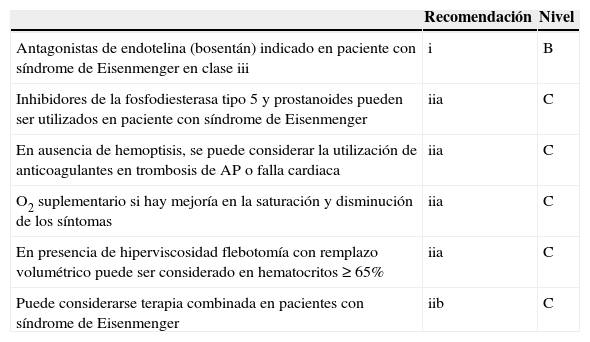
Nivel de recomendación hipertensión pulmonar secundaria a cardiopatía congénita
| Recomendación | Nivel | |
|---|---|---|
| Antagonistas de endotelina (bosentán) indicado en paciente con síndrome de Eisenmenger en clase iii | i | B |
| Inhibidores de la fosfodiesterasa tipo 5 y prostanoides pueden ser utilizados en paciente con síndrome de Eisenmenger | iia | C |
| En ausencia de hemoptisis, se puede considerar la utilización de anticoagulantes en trombosis de AP o falla cardiaca | iia | C |
| O2 suplementario si hay mejoría en la saturación y disminución de los síntomas | iia | C |
| En presencia de hiperviscosidad flebotomía con remplazo volumétrico puede ser considerado en hematocritos≥65% | iia | C |
| Puede considerarse terapia combinada en pacientes con síndrome de Eisenmenger | iib | C |
Modificada de Galié et al.19.
Teniendo en cuenta que aproximadamente el 20% de los pacientes con síndrome de Eisenmenger presentan eventos trombóticos o tromboembólicos en la circulación pulmonar y dichos cambios se correlacionan con la edad, disfunción ventricular, dilatación del tronco y ramas de la arteria pulmonar, parecería razonable el uso de anticoagulación profiláctica. Sin embargo, la utilización de anticoagulantes o antiagregantes plaquetarios se relaciona con un mayor riesgo de hemorragia, por lo que su uso está restringido a pacientes con taquiarritmias supraventriculares, con prótesis mecánica o con evidencia de trombosis arterial pulmonar sin hemoptisis y en fallo cardiaco70,71. Un estudio observacional reciente muestra que los pacientes con síndrome de Eisenmenger que son anticoagulados de manera rutinaria no tienen una mayor supervivencia72.
Está bien establecido que el embarazo en pacientes con síndrome de Eisenmenger se asocia con una elevada incidencia de aborto (de hasta un 40%), partos prematuros en la mitad de los casos y una mortalidad materna del 50%. La mayoría de las muertes maternas ocurren en el periodo periparto durante el alumbramiento o en la primera semana posparto. Sin embargo, los efectos del embarazo en el área cardiovascular persisten durante meses después del parto por lo que no es excepcional observar muertes tardías. Se piensa que el reto hemodinámico primordial ocurre durante el parto ya que la hipercapnia y la acidosis elevan la resistencia vascular pulmonar lo que conduce a una insuficiencia cardiaca derecha grave. Si bien con el mejor conocimiento de la fisiopatología cardiopulmonar y la utilización de diversos medicamentos en la vasculatura pulmonar y la conformación de equipos multidisciplinarios se ha logrado reducir la mortalidad materna hasta un 28%, sigue siendo prohibitiva, por lo que es primordial insistir a las pacientes sobre la necesidad de evitar el embarazo así como contar con métodos anticonceptivos adecuados. Deben evitarse métodos de barrera como método de anticoncepción por su elevado índice de fallo y el uso de anticonceptivos que contengan estrógenos por la propensión a eventos tromboembólicos. Debido a lo anterior son de elección la salpingoclasia o la esterilización de la pareja. La terminación de la gestación en los 2 últimos trimestres del embarazo tiene un alto riesgo para la madre, por lo que se deberá analizar los riesgos y beneficios en cada uno de los casos73,74.
Si bien en un estudio que se desarrolló en 32 pacientes adultos con hipertensión pulmonar sometidos a 15 semanas de entrenamiento aeróbico de baja intensidad se demostró que mejoraron de manera significativa la tolerancia al ejercicio, su beneficio no ha sido aún evaluado en los niños75,76.
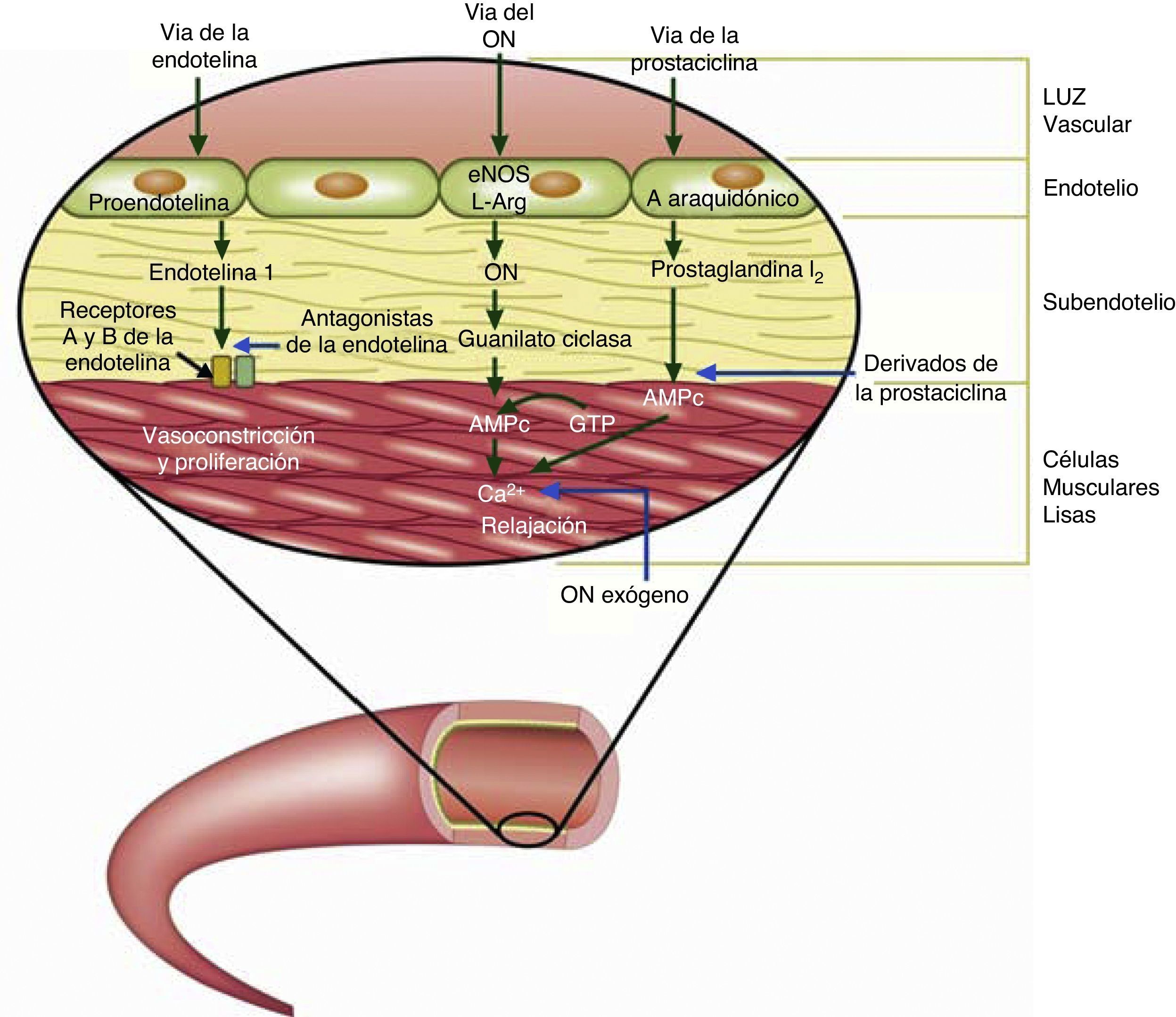
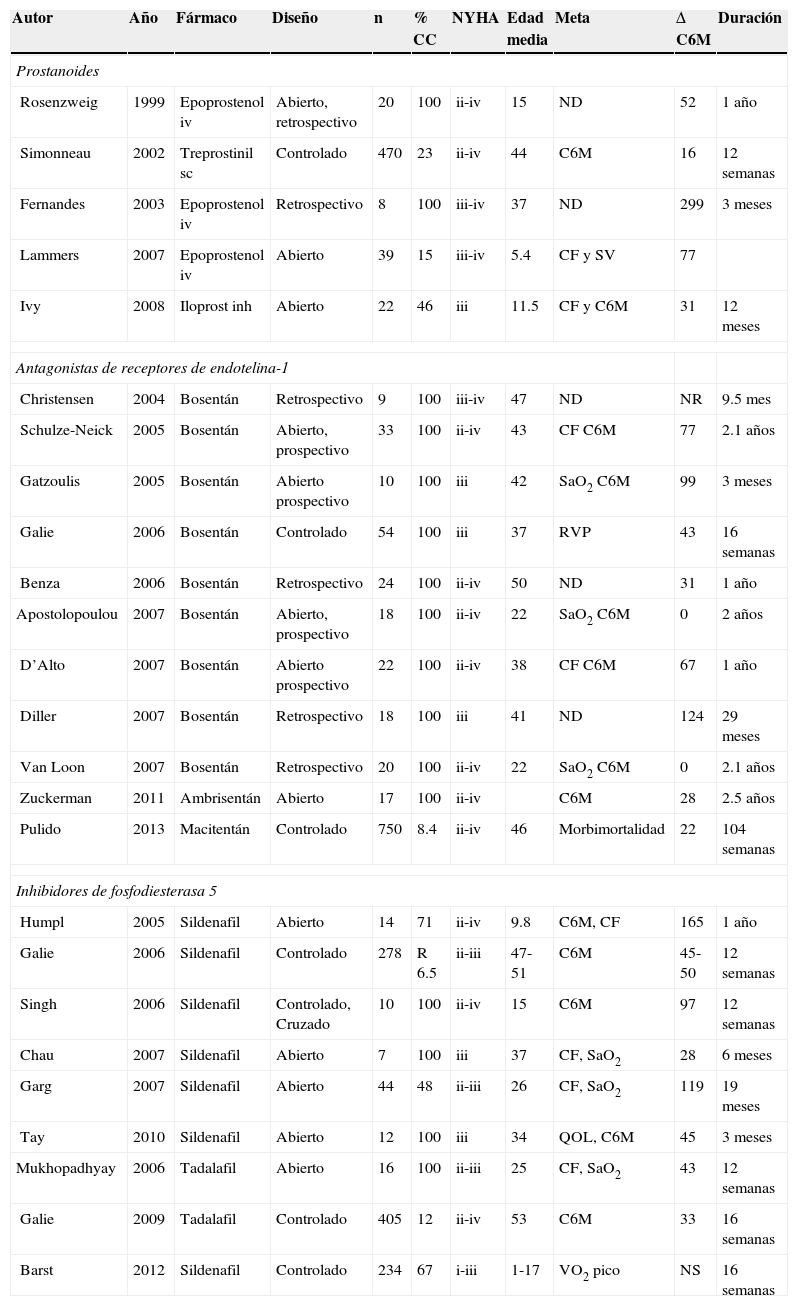
Tratamiento con fármacos específicos para hipertensión pulmonarEn el tratamiento de la HAP asociada a CC, los fármacos empleados han intentado modificar las 3 vías fisiopatológicas conocidas (prostanoides, antagonistas de receptores de endotelina-1 y los inhibidores de la fosfodiesterasa 5 (fig. 4). Existen pocos estudios controlados orientados específicamente a esta población (tabla 8) y en ellos la etiología en cuanto a la CC de base es variable. Existen asimismo múltiples series abiertas (no controladas),en las que las dosis de los fármacos son diferentes, al igual que los objetivos primarios para evaluar eficacia. Los diferentes estudios realizados varían también en duración. Por todo lo anterior, las recomendaciones en cuanto al empleo de estos fármacos son extrapoladas de su uso en la población general de HAP, asumiendo en su mayor parte efectos similares.

Algunos estudios relevantes en el tratamiento de la HAP asociada a cardiopatía congénita
| Autor | Año | Fármaco | Diseño | n | % CC | NYHA | Edad media | Meta | Δ C6M | Duración |
|---|---|---|---|---|---|---|---|---|---|---|
| Prostanoides | ||||||||||
| Rosenzweig | 1999 | Epoprostenol iv | Abierto, retrospectivo | 20 | 100 | ii-iv | 15 | ND | 52 | 1 año |
| Simonneau | 2002 | Treprostinil sc | Controlado | 470 | 23 | ii-iv | 44 | C6M | 16 | 12 semanas |
| Fernandes | 2003 | Epoprostenol iv | Retrospectivo | 8 | 100 | iii-iv | 37 | ND | 299 | 3 meses |
| Lammers | 2007 | Epoprostenol iv | Abierto | 39 | 15 | iii-iv | 5.4 | CF y SV | 77 | |
| Ivy | 2008 | Iloprost inh | Abierto | 22 | 46 | iii | 11.5 | CF y C6M | 31 | 12 meses |
| Antagonistas de receptores de endotelina-1 | ||||||||||
| Christensen | 2004 | Bosentán | Retrospectivo | 9 | 100 | iii-iv | 47 | ND | NR | 9.5 mes |
| Schulze-Neick | 2005 | Bosentán | Abierto, prospectivo | 33 | 100 | ii-iv | 43 | CF C6M | 77 | 2.1 años |
| Gatzoulis | 2005 | Bosentán | Abierto prospectivo | 10 | 100 | iii | 42 | SaO2 C6M | 99 | 3 meses |
| Galie | 2006 | Bosentán | Controlado | 54 | 100 | iii | 37 | RVP | 43 | 16 semanas |
| Benza | 2006 | Bosentán | Retrospectivo | 24 | 100 | ii-iv | 50 | ND | 31 | 1 año |
| Apostolopoulou | 2007 | Bosentán | Abierto, prospectivo | 18 | 100 | ii-iv | 22 | SaO2 C6M | 0 | 2 años |
| D’Alto | 2007 | Bosentán | Abierto prospectivo | 22 | 100 | ii-iv | 38 | CF C6M | 67 | 1 año |
| Diller | 2007 | Bosentán | Retrospectivo | 18 | 100 | iii | 41 | ND | 124 | 29 meses |
| Van Loon | 2007 | Bosentán | Retrospectivo | 20 | 100 | ii-iv | 22 | SaO2 C6M | 0 | 2.1 años |
| Zuckerman | 2011 | Ambrisentán | Abierto | 17 | 100 | ii-iv | C6M | 28 | 2.5 años | |
| Pulido | 2013 | Macitentán | Controlado | 750 | 8.4 | ii-iv | 46 | Morbimortalidad | 22 | 104 semanas |
| Inhibidores de fosfodiesterasa 5 | ||||||||||
| Humpl | 2005 | Sildenafil | Abierto | 14 | 71 | ii-iv | 9.8 | C6M, CF | 165 | 1 año |
| Galie | 2006 | Sildenafil | Controlado | 278 | R 6.5 | ii-iii | 47-51 | C6M | 45-50 | 12 semanas |
| Singh | 2006 | Sildenafil | Controlado, Cruzado | 10 | 100 | ii-iv | 15 | C6M | 97 | 12 semanas |
| Chau | 2007 | Sildenafil | Abierto | 7 | 100 | iii | 37 | CF, SaO2 | 28 | 6 meses |
| Garg | 2007 | Sildenafil | Abierto | 44 | 48 | ii-iii | 26 | CF, SaO2 | 119 | 19 meses |
| Tay | 2010 | Sildenafil | Abierto | 12 | 100 | iii | 34 | QOL, C6M | 45 | 3 meses |
| Mukhopadhyay | 2006 | Tadalafil | Abierto | 16 | 100 | ii-iii | 25 | CF, SaO2 | 43 | 12 semanas |
| Galie | 2009 | Tadalafil | Controlado | 405 | 12 | ii-iv | 53 | C6M | 33 | 16 semanas |
| Barst | 2012 | Sildenafil | Controlado | 234 | 67 | i-iii | 1-17 | VO2 pico | NS | 16 semanas |
Fuente: referencias 83-110.
CF: clase funcional; C6M: caminata de los 6min; %CC: porcentaje de pacientes con cardiopatía congénita; inh: inhalado; iv: intravenoso; ND: no disponible; NR: no reportada; NS: no significativa; NYHA: New York Heart Association; QOL: calidad de vida; RVP: resistencia vascular pulmonar; SaO2%: saturación arterial de oxígeno; sc: subcutáneo; SV: supervivencia; VO2: consumo de oxígeno.
Las guías de la Sociedad Europea de Cardiología recomiendan en los pacientes con síndrome de Eisenmenger en clase funcional iii la utilización de bosentán, antagonista no selectivo de los receptores de endotelina, sustentado en el primer estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo el Bosentan Randomised Trial of Endothelin Antagonist THerapy (BREATHE-5) en el que se evaluó durante 16 semanas la saturación de oxígeno, aspectos hemodinámicos y la capacidad de ejercicio, en pacientes con síndrome de Eisenmenger. Los resultados iniciales mostraron una disminución de la presión media pulmonar, mejoría en el índice de resistencia vascular pulmonar e incremento en la capacidad de ejercicio; la prolongación del estudio a 24 semanas mostró que dichos beneficios se mantenían77,78. Posteriormente, diferentes investigaciones han confirmado esta respuesta 79–80(tablas 7 y 8).
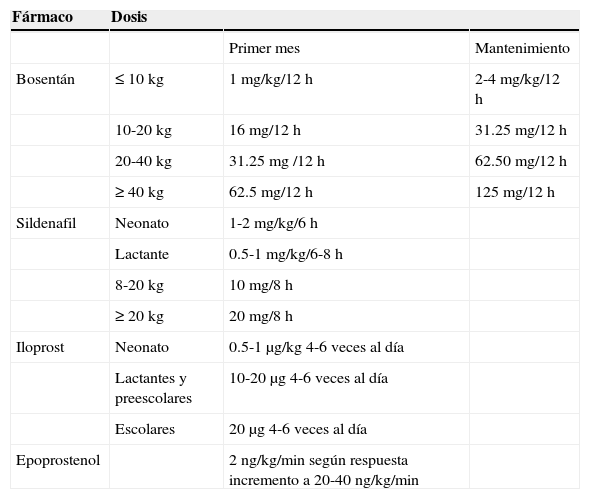
En cuanto a los inhibidores de la fosfodiesterasa 5, tanto el sildenafil como el tadalafil han mostrado mejorar la capacidad funcional y al esfuerzo, el score de Borg y aspectos hemodinámicos en pacientes con HAP secundaria a CC y síndrome de Eisenmenger. Los estudios START-1 y su extensión START-2 fueron multicéntricos, doble ciego, aleatorizados, controlados con placebo: Niños con HAP (1 a 17 años de edad con peso>8kg) recibieron una dosis baja de 10mg, dosis media de 10-40mg o alta de 20-80mg 3 veces al día. La razón de momios para mortalidad fue 3.95 (1.46-10.65) al relacionar dosis alta en comparación con dosis baja y de 1.92 (0.65-5.65) para dosis media a baja. Tomando como base esta información se ha recomendado la dosis de 10mg 3 veces al día en pacientes con peso<20kg y de 20mg en pacientes con peso>20kg81–85 (tabla 9) En agosto del 2012 la FDA alertó sobre el uso crónico del sildenafil en pacientes pediátricos<17 años82.
Medicamentos vasoactivos para el manejo de la HAP
| Fármaco | Dosis | ||
|---|---|---|---|
| Primer mes | Mantenimiento | ||
| Bosentán | ≤10kg | 1mg/kg/12h | 2-4mg/kg/12h |
| 10-20kg | 16mg/12h | 31.25mg/12h | |
| 20-40kg | 31.25mg /12h | 62.50mg/12h | |
| ≥40kg | 62.5mg/12h | 125mg/12h | |
| Sildenafil | Neonato | 1-2mg/kg/6h | |
| Lactante | 0.5-1mg/kg/6-8h | ||
| 8-20kg | 10mg/8h | ||
| ≥20kg | 20mg/8h | ||
| Iloprost | Neonato | 0.5-1μg/kg 4-6 veces al día | |
| Lactantes y preescolares | 10-20μg 4-6 veces al día | ||
| Escolares | 20 μg4-6 veces al día | ||
| Epoprostenol | 2ng/kg/min según respuesta incremento a 20-40ng/kg/min |
Diversas publicaciones describen el beneficio de estas terapias vasodilatadoras. En un estudio retrospectivo que incluyó a 229 pacientes con síndrome de Eisenmenger, el uso de diversos fármacos con efecto vasodilatador pulmonar (bosentán 73.5%; sildenafil 25% y epoprostenol 1.5%) con un promedio de seguimiento de 4 años se asoció con una disminución significativa de la mortalidad al compararlo con aquellos que no la recibieron (2 versus 52 pacientes)86. Aunque la evidencia es limitada, en aquellos pacientes sintomáticos o con deterioro clínico y que han sido tratados con una monoterapia vasodilatadora, debe ser considerada la asociación de diversos fármacos buscando un efecto sinérgico, destacando la asociación de sildenafil y bosentán. En el registro REVEAL pediátrico10, el 37% recibían monoterapia mientras que el 29% tuvieron doble o triple combinación con medicación específica, algo difícilmente alcanzable en nuestro medio87–111.
En la práctica clínica la alternativa del trasplante está restringida a pacientes muy sintomáticos con una expectativa de vida corta. En pacientes con síncope, insuficiencia cardiaca derecha refractaria, clase funcional iv (NYHA) e hipoxia grave puede considerarse la opción de trasplante pulmonar con corrección de la malformación cardiaca o trasplante corazón-pulmón. Las tasas de supervivencia a un año son del 78% y disminuyen al 49% a 5 años y al 25% a 10 años112–114.
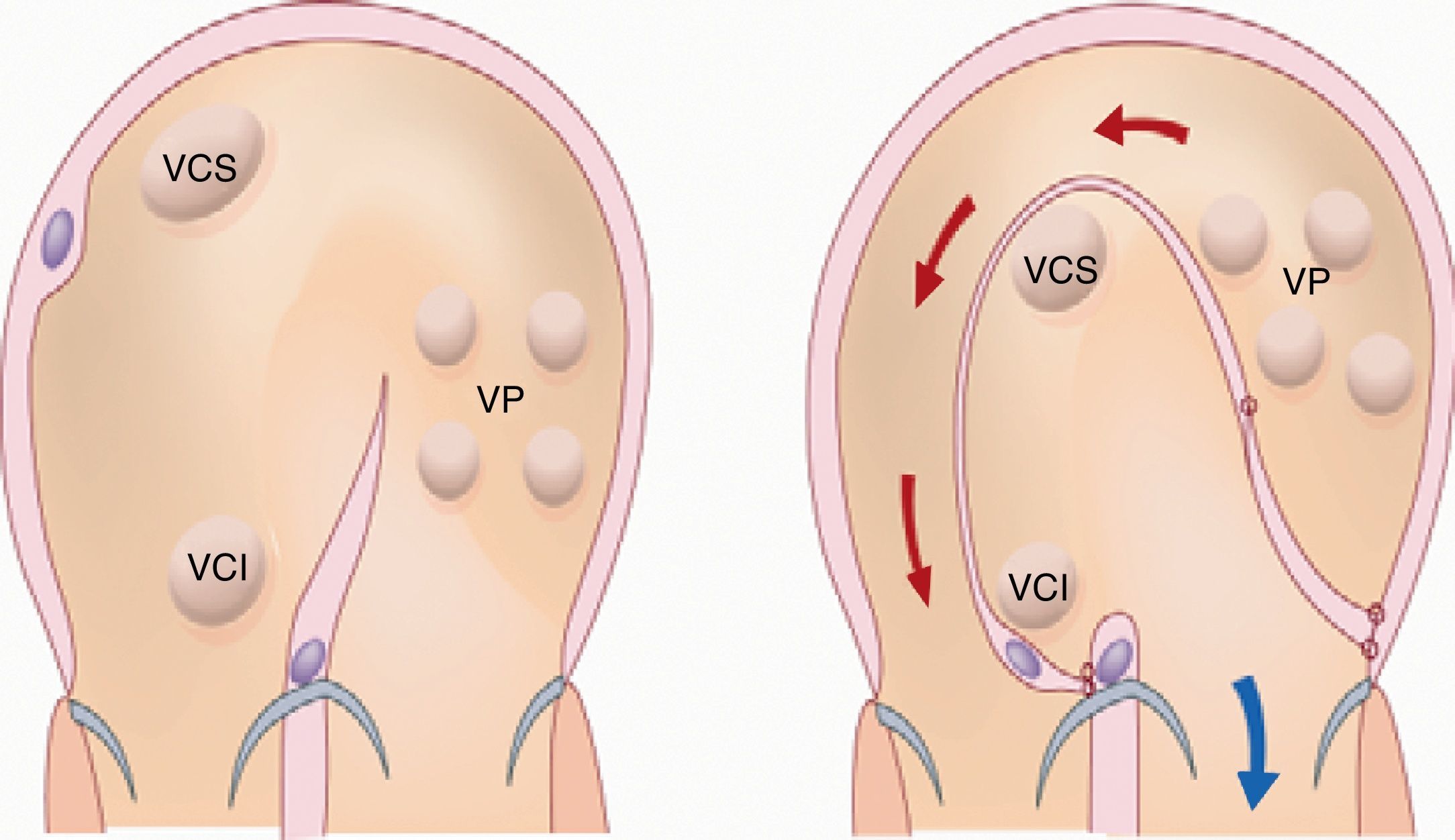
Otras terapéuticasLa realización de la cirugía de Mustard es un planteamiento interesante por los beneficios que derivan de la reducción de la hipoxia en paciente con HAP secundaria a CC (fig. 2). En pacientes con trasposición de grandes arterias con una comunicación interventricular grande, la realización de esta cirugía, de redirección del flujo, sin llevar a cabo cierre de la comunicación interventricular incrementa la saturación de oxígeno en la arteria pulmonar y clínicamente disminuye la sintomatología y mejora la calidad de vida115,116 (fig. 5). Es importante señalar que la hipoxia presente en los pacientes con CC complejas contribuye a una mayor mineralización vascular, manifestación de disfunción endotelial117.

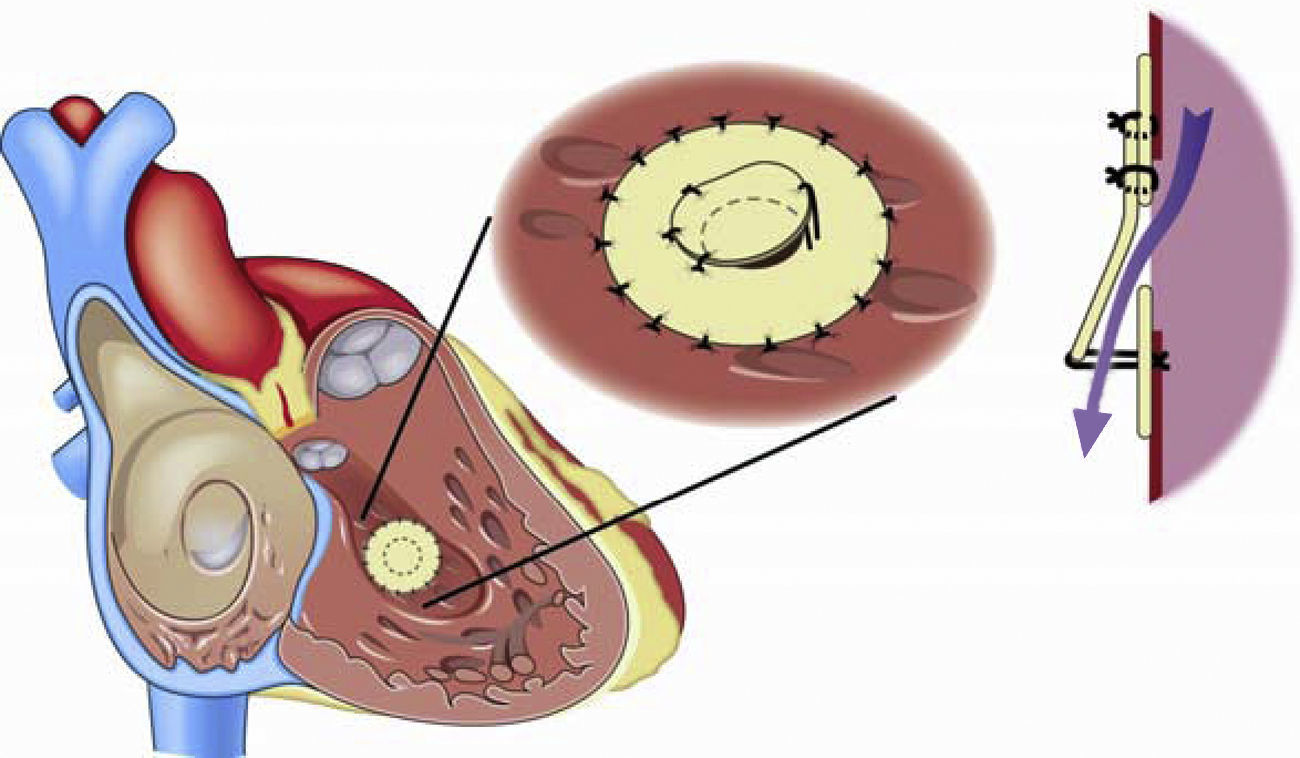
La respuesta a terapias vasodilatadoras en los pacientes con síndrome de Eisenmenger en clase funcional i-ii (NYHA) es un campo que requiere ser estudiado; hasta el momento la respuesta no es uniforme ya que algunos pacientes muestran un beneficio importante, mientras otros mínimo o ningún cambio. El llevar a cabo tratamiento con terapia vasodilatadora con un seguimiento puntual de los aspectos clínicos y hemodinámicos es una alternativa muy recomendable antes de proceder al cierre de cortocircuitos intracardiacos118. Asimismo, el cierre parcial de defectos o el uso de un parche valvulado fenestrado (fig. 6) que permita un cortocircuito de derecha a izquierda a fin de descargar el ventrículo derecho en episodios de crisis hipertensiva pulmonar puede ser una alternativa, en especial de utilidad durante el periodo postoperatorio119.

Existen algunos informes recientes que hablan del beneficio de llevar a cabo bandaje pulmonar en pacientes HAP avanzada. Batista et al.120 informan sobre un paciente con comunicación interauricular e interventricular con HAP grave en quien fue posible hacer corrección total un año después de llevar a cabo el bandaje de la arteria pulmonar. Por otra parte, Khan et al. reportan los casos de 4 niños entre 3 y 10 años de edad con comunicación interventricular grande y HAP grave que fueron sometidos a bandaje de la arteria pulmonar; 2 de ellos tuvieron una buena evolución a medio plazo, uno falleció y al otro se le inició tratamiento con bosentán121.
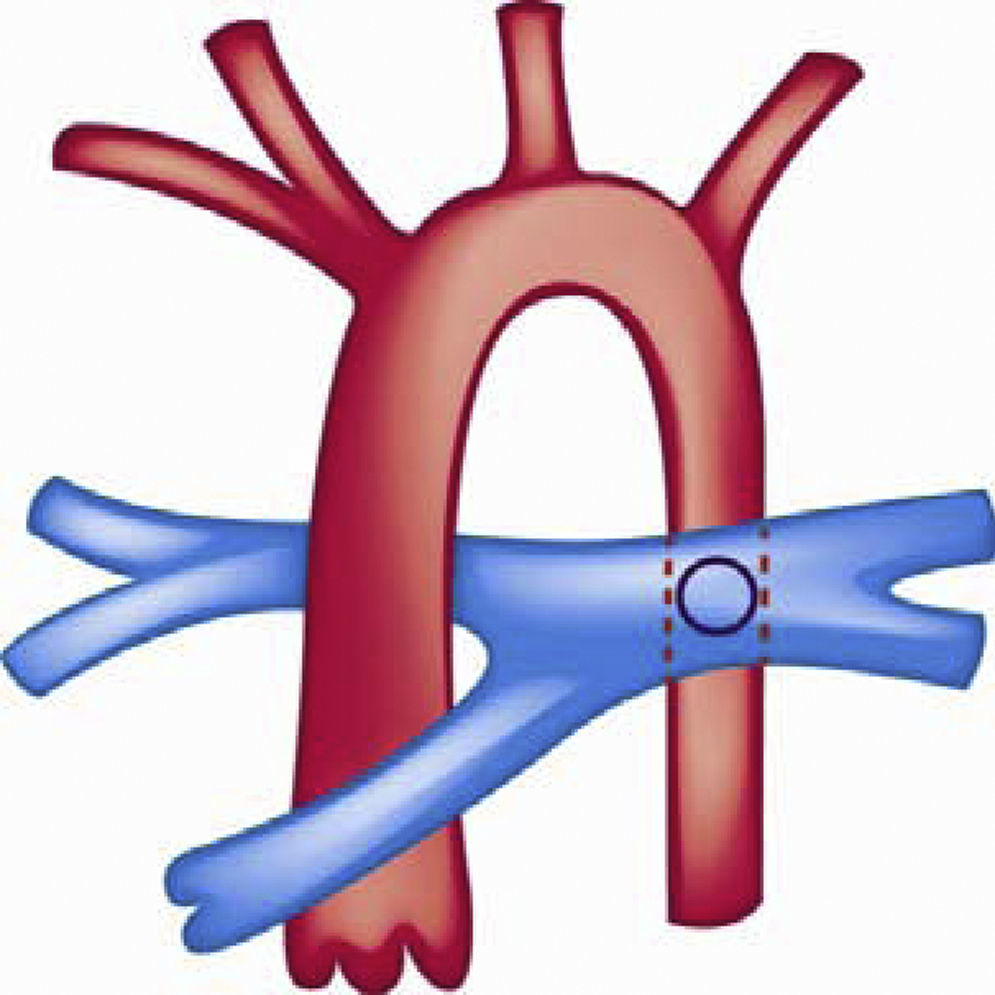
Se han utilizado otras alternativas quirúrgicas como la realización de una fistula sistémico-pulmonar tipo Potts que consiste en una anastomosis entre la rama pulmonar izquierda y la aorta descendente en pacientes que fueron llevados a corrección de su CC pero que finalmente desarrollaron hipertensión pulmonar grave y tenían insuficiencia cardiaca derecha (fig. 7). Dicha cirugía se ha utilizado como alternativa para mejorar el gasto sistémico y el estado clínico del paciente122,123.
Síndrome de Down e hipertensión pulmonar
La susceptibilidad del paciente con síndrome de Down para desarrollar hipertensión pulmonar continúa siendo una incógnita. En 1866, Down describe las características clínicas del síndrome que ahora lleva su nombre; ya en 1959 Lejeune 124 y Jacobs et al.125, de manera independiente, determinan que dicho fenotipo era consecuencia a una trisomía del cromosoma 21. Se estima una incidencia aproximada de 1.1 por 1,000 nacidos vivos126,127. El síndrome de Down es una cromosomopatía bien caracterizada asociada a diversas morbilidades dentro de las que destacan las CC, que se presentan en aproximadamente el 50% de los pacientes y son la principal causa de mortalidad en este síndrome. El tipo de cardiopatía más frecuente varía de acuerdo a la serie reportada; sin embargo, las principales son: la persistencia del conducto arterioso, el defecto de la tabicación auriculoventricular y la comunicación interventricular128. Está bien establecido que una de las principales complicaciones de las CC es el desarrollo de HAP. Algunos estudios refieren que los pacientes con síndrome de Down y CC tienen una inusual elevación de las resistencias vasculares pulmonares y una propensión al desarrollo de daño precoz y severo del lecho vascular pulmonar. También se menciona que la progresión de la HAP es decurso más rápido en niños con síndrome de Down con CC y se estima que, aproximadamente, la mitad de todos los niños con defectos septales interventriculares grandes pueden desarrollar enfermedad vascular pulmonar alrededor de los 2 años de vida. El desarrollo temprano de HAP se atribuye a disfunción endotelial. La exposición constante de la vasculatura pulmonar a un mayor flujo y presión condicionan remodelación vascular y disfunción endotelial, lo que a su vez provoca un incremento en la resistencia vascular pulmonar y finalmente revierte el cortocircuito y da lugar al síndrome de Eisenmenger129–136. La prevalencia de síndrome de Down en el TOPP9 fue del 13% y en el registro Holandés8 fue del 12%. En un estudio que realizamos en nuestra Institución cuyo objetivo fue describir el comportamiento de la HAP grave en pacientes con síndrome de Down operados de comunicación interventricular y compararlos con aquellos no sindrómicos para determinar si el hecho de ser portador de síndrome de Down conlleva mayor riesgo de hipertensión pulmonar persistente, se encontró que en un seguimiento medio de 554±518 días, la presión pulmonar media medida por ecocardiografía en los pacientes con síndrome de Down fue de 44.8±15.1 versus 35.1±12.1mmHg en los no sindrómicos con un valor de p≥0.0275137.
ConclusionesLa enfermedad vascular pulmonar consecutiva a CC es la etiología más prevenible de hipertensión arteria pulmonar. Sin embargo, dada la alta prevalencia de las CC aunada a una insuficiente cobertura para su detección, estudio y tratamiento oportuno en nuestro país, la hipertensión pulmonar es y será una consecuencia a la que nos tendremos que seguir enfrentando. Por todo lo anterior es indispensable tener un conocimiento fundamentado de las posibilidades de abordaje y de las diversas estrategias terapéuticas con el objetivo de ofrecer las mejores opciones.
FinanciaciónNo se recibió patrocinio de ningún tipo para la escritura de este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.