




La hipertensión portopulmonar (HPP) es una entidad poco frecuente a nivel mundial, aunque se desconocen los datos epidemiológicos en México. Sin embargo, las enfermedades crónicas del hígado son muy prevalentes en mexicanos. La HPP es el 4.° subtipo en frecuencia del grupo de la hipertensión arterial pulmonar. Su diagnóstico está dentro de 2 escenarios: los pacientes con sospecha de hipertensión pulmonar y los candidatos a trasplante hepático ortotópico (THO). Tanto el ecocardiograma como el cateterismo cardiaco derecho son determinantes para el diagnóstico en ambos escenarios. La HPP es un reto para el THO, pues aumenta la mortalidad perioperatoria de manera importante. El uso de terapia específica es la piedra angular de este padecimiento, como una medida para poder mejorar el desenlace de los que llegan a ser candidatos a un THO con HPP moderada a grave. Es importante reconocer que la HPP puede llegar a ser una contraindicación para el THO. Hasta el momento el papel del trasplante combinado pulmón-hígado o corazón-pulmón-hígado como una medida de curación de la enfermedad vascular pulmonar en pacientes con HPP es incierto.
Portopulmonary hypertension (PPH) is a rare condition worldwide, although epidemiological data are unknown in Mexico. However, chronic liver diseases are very prevalent in Mexico. PPH is the 4th subtype in frequency in the group of pulmonary arterial hypertension. Its diagnosis is made within 2 scenarios: patients with suspected pulmonary hypertension and candidates for orthotopic liver transplantation (OLT). Both echocardiogram and a right cardiac catheterisation are crucial for diagnosis in both cases. PPH is a challenge for OLT, since it can significantly increase perioperative mortality. The use of specific therapy is the cornerstone of this disease, as a measure to improve the outcome of those who become candidates for OLT with moderate to severe PPH. It is important to recognise that PPH can be a contraindication to OLT. The role of lung-liver transplantation or heart-lung-liver transplantation as a measure to heal pulmonary vascular disease in patients with PPH is still uncertain.
El hígado en condiciones estables recibe el 15-20% del gasto cardiaco, de los cuales 2 terceras partes provienen del sistema venoso portal (circuito de baja presión)1. La hipertensión portal (HPo) es común y se caracteriza por un aumento en el gradiente de presión entre la vena porta y la cava inferior2, lo que hemodinámicamente se define por un gradiente de presión venosa hepática (GPVH)≥6mm Hg, una presión transesplénica≥15mm Hg y/o una presión de la vena porta≥21mm Hg1. Su etiología puede variar a lo largo del tiempo, la edad y estado socioeconómico de la población de un país3, sin embargo, la causa más frecuente es la cirrosis hepática (CH) o HPo hepática2. Se estima que en Occidente corresponde al 88-90% de los casos, mientras que en el Lejano Oriente al 70%4–6.
La enfermedad hepática crónica y la ascendenciaHasta el momento lo que conocemos en México de la hipertensión portopulmonar (HPP) es solo información proveniente de países desarrollados, cuyo control en enfermedades crónicas del hígado son eficientes.
En 2010, el 2% del total de muertes en el mundo estuvo relacionada con la CH, lo que representa un aumento bruto del 0.4% desde 1980, pero una disminución del 21.6% de la tasa estandarizada por edad en estos últimos 30 años. México no está exento de esta disminución (28.3%), sin embargo, la tasa de mortalidad (38.3/100,000 habitantes en 2010) es la más alta de América Latina y la octava a nivel mundial (Egipto es el número uno con 72.7/100,000 habitantes)7.
A pesar del mestizaje, resultado de la Conquista de América, en España las enfermedades crónicas del hígado se mantuvieron como la causa número 11 de mortalidad en el 2011, con cerca del 1.2% del porcentaje total de defunciones8. Independientemente del crecimiento de la población hispana en los Estados Unidos de América (EUA) (54 millones en 2013 con un 64% de ascendencia mexicana)9, la CH y las enfermedades crónicas del hígado no estuvieron dentro de las 10 primeras causas de muerte en 2010 en la población general, y al realizar el análisis por grupos étnicos de ascendencia, se encontraron cambios en la mortalidad por dichas enfermedades; por ejemplo, el total de muertes en afroamericanos fue del 0.9%, de blancos no hispanos fue del 1.2%, en nativos americanos del 5.1% y en hispanos del 3%10. Existe información que hace diferente el área conocida como la zona fronteriza mexicoestadounidense (100km de ancho a cada lado de la frontera), de tal forma que en el lado estadounidense por lo menos el 50% de la población es hispana, y de estos mayormente descendientes de mexicanos, en esta población las enfermedades crónicas del hígado en 2007 fueron la novena causa de mortalidad (10.6-18.9/100, 000 habitantes), mientras en el lado mexicano fueron la séptima causa (17.6-29.8/100,000 habitantes)11.
En México de acuerdo al Instituto Nacional de Estadística y Geografía, la CH y otras enfermedades crónicas del hígado representan la quinta causa de muerte en la población general desde el 2000 hasta el 2013 (un 6.3% del total en 2000, un 5.5% en 2010 y un 5.5% en 2013). De acuerdo a la distribución por género, en hombres representa la 5.a causa, y en mujeres entre la 7.a y 6.a causa, de acuerdo al año analizado. Por grupos de edad, de 35 a 44 años pasó de ser la 2.acausa en el 2000 a la 4.a causa en el año 2013. En el grupo de 45 a 64 años representa la 4.a causa de mortalidad, en este mismo grupo es la 3.a causa en hombres y la 4.a en mujeres. Si se realiza un análisis combinado, en el grupo de 35 a 44 años, vemos que para el 2000, el 6.5% de las muertes en mujeres fue por enfermedades crónicas del hígado, para el 2010 fue del 4.9% y para el 2013 fue del 4.7%. Mientras que, en hombres dentro del mismo grupo etario, representó el 18.8, 12.4 y 13.8% para los años 2000, 2010 y 2013 respectivamente. Una tendencia similar se aprecia en el grupo de 45 a 64 años, donde en mujeres para el 2000 la mortalidad fue del 6.9%, en 2010 fue del 6% y en 2013 fue del 6.1%, mientras que en hombres se aprecian cifras del 17.2% (2000), 14% (2010) y 14% (2013). De acuerdo a estos datos, la CH es un verdadero problema de salud en México12.
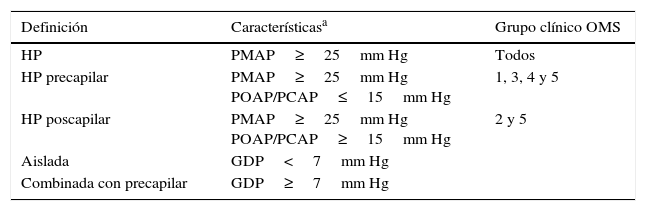
Conceptos en relación con hipertensión portopulmonarEs importante diferenciar la hipertensión pulmonar (HP), la hipertensión arterial pulmonar (HAP) y la HPP. Así, la HP es un aumento de la presión media de la arteria pulmonar (PMAP), mientras que la HAP es un subtipo de HP y corresponde al grupo 1 de la Organización Mundial de la Salud (OMS), mientras la HPP es parte del grupo de HAP (tabla 1). No está de más recalcar que la definición de HP es hemodinámica (tabla 2), y para identificar a los pacientes con HAP se deben de descartar otras causas de HP apoyándose en datos clínicos y auxiliares diagnósticos que sigan un flujograma bien establecido13. La importancia de identificar a los pacientes con HAP radica en que es una entidad que condiciona un deterioro progresivo tanto de la circulación pulmonar (remodelación arterial), como del corazón (falla ventricular derecha)14.
Definición hemodinámica de hipertensión pulmonar
| Definición | Característicasa | Grupo clínico OMS |
|---|---|---|
| HP | PMAP≥25mm Hg | Todos |
| HP precapilar | PMAP≥25mm Hg POAP/PCAP≤15mm Hg | 1, 3, 4 y 5 |
| HP poscapilar | PMAP≥25mm Hg POAP/PCAP≥15mm Hg | 2 y 5 |
| Aislada | GDP<7mm Hg | |
| Combinada con precapilar | GDP≥7mm Hg |
GDP: gradiente diastólico de presión pulmonar (=PDAP−POAP/PCAP); HP: hipertensión pulmonar; PDAP: presión diastólica de la arteria pulmonar; PMAP: presión media de la arteria pulmonar; POAP/PCAP: presión de oclusión de la arteria pulmonar/presión en cuña de la arteria pulmonar.
Adaptada de Galiè et al.13.
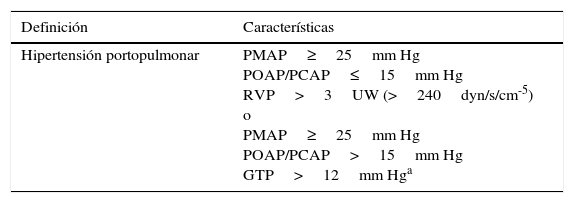
Definición y clasificación de la hipertensión portopulmonar
| Definición | Características |
|---|---|
| Hipertensión portopulmonar | PMAP≥25mm Hg POAP/PCAP≤15mm Hg RVP>3UW (>240dyn/s/cm-5) o PMAP≥25mm Hg POAP/PCAP>15mm Hg GTP>12mm Hga |
| Clasificación | PMAP |
|---|---|
| Leve | PMAP≥25-34mm Hg |
| Moderada | PMAP35-44mm Hg |
| Grave | PMAP≥45mm Hg |
GTP: gradiente transpulmonar (=PMAP–POAP/PCAP); PMAP: presión media de la arteria pulmonar; POAP/PCAP: presión de oclusión de la arteria pulmonar/presión en cuña de la arteria pulmonar; RVP: resistencia vascular pulmonar.
Adaptada de Krowka y Rodriguez-Roisin27.
Los pacientes con CH se pueden presentar con HP relacionada con el estado hiperdinámico característico, con sobrecarga de volumen y con eventos tromboembólico venosos recurrentes15. Sin embargo, la asociación de HPo clínica o hemodinámica e HAP con o sin afección hepática crónica se conoce como HPP13, y no está relacionada con la etiología de la enfermedad hepática, ni con la gravedad de la HPo (tabla 3)16. Los primeros en describir esta asociación fueron Mantz y Craig en 195115, y hasta antes de 1998 estaba comprendida en el grupo de etiologías «secundarias» de la HP, mientras que ahora está incluida en el grupo 1 de la OMS, mejor conocido como HAP (tabla 1)13.
Clasificación actualizada de la hipertensión pulmonar
| 1. Hipertensión arterial pulmonar |
| 1.1 Hipertensión arterial pulmonar idiopática |
| 1.2 Hipertensión arterial pulmonar hereditaria |
| 1.2.1 BMPR2 |
| 1.2.2 ALK-1, ENG, SMAD9, CAV1, KCNK3 |
| 1.2.3 Desconocido |
| 1.3 Inducida por fármacos y toxinas |
| 1.4. Asociada con |
| 1.4.1 Enfermedad del tejido conectivo |
| 1.4.2 Infección por el VIH |
| 1.4.3 Hipertensión portal |
| 1.4.4 Enfermedad cardiaca congénita |
| 1.4.5 Esquistosomiasis |
| 1′ Enfermedad venooclusiva pulmonar y/o hemangiomatosis capilar pulmonar |
| 1′’ Hipertensión pulmonar persistente del recién nacido |
| 2. Hipertensión pulmonar debida a enfermedad del corazón izquierdo |
| 2.1. Disfunción sistólica del ventrículo izquierdo |
| 2.2 Disfunción diastólica del ventrículo izquierdo |
| 2.3. Enfermedad valvular |
| 2.4 Obstrucción del tracto de entrada/salida del corazón izquierdo congénita/adquirida y miocardiopatías congénitas |
| 3. Hipertensión pulmonar debida a enfermedades pulmonares y/o hipoxia |
| 3.1 Enfermedad pulmonar obstructiva crónica |
| 3.2 Neumopatía intersticial |
| 3.3 Otras neumopatías con patrón mixto restrictivo y obstructivo |
| 3.4 Desórdenes respiratorios del sueño |
| 3.5 Desórdenes de hipoventilación alveolar |
| 3.6 Exposición crónica a elevada altitud |
| 3.7 Enfermedades del desarrollo pulmonar |
| 4. Hipertensión pulmonar tromboembólica crónica |
| 5. Hipertensión pulmonar con mecanismos multifactoriales poco claros |
| 5.1 Desórdenes hematológicos: anemia hemolítica crónica, desórdenes mieloproliferativos, esplenectomía |
| 5.2. Desórdenes sistémicos: sarcoidosis, histiocitosis pulmonar, linfangioleiomiomatosis |
| 5.3 Desórdenes metabólicos: enfermedades del almacenamiento del glucógeno, enfermedad de Gaucher, desórdenes tiroideos |
| 5.4 Otros: obstrucción tumoral, mediastinitis fibrosante, enfermedad renal crónica, hipertensión pulmonar segmentaria |
Adaptada de Galiè et al.13.
Del grupo 1 de la OMS para HP, los 4 subtipos más frecuentes son la HAP idiopática (HAPI) con el 39-61% de los casos, las formas de HAP asociada a enfermedades del tejido conectivo (11-28%), a cardiopatías congénitas (10-20%) y a HPo (5-10%). La edad de diagnóstico de la HAPI en la mayoría de los registros es en la 6.a década de la vida17–19, mientras que los casos de HPP se diagnostican alrededor de los 50 años4,20, aunque aquellos con etiología no hepática de la HPo son de menor edad (±40 años de edad)8. En el registro REVEAL, la relación entre mujeres y hombres en HPP es 1:120, aunque en los casos secundarios a enfermedad hepática autoinmune el predominio es en el género femenino21.
En 17,901 autopsias en pacientes con un año de edad o más, el 6.9% tuvieron CH, y en el 0.73% de aquellos con CH se detectaron cambios histopatológicos compatibles con HAP22. Dentro del contexto de la HPP las etiologías más comunes de la CH son por alcohol4, virus de hepatitis C y hepatitis autoinmune21, pero a pesar de que la HPP con HPo no hepática es infrecuente, la trombosis portal4 y la HPo idiopática6 son las causas más importantes.
En los pacientes con HPo no importa la gravedad de la misma, pues a mayor tiempo de evolución (4-7 años en promedio) la probabilidad de desarrollar HPP se incrementa15,21,23. En el caso de los pacientes con una derivación portosistémica quirúrgica, el 65% padecían HPP24 sin embargo, el tiempo entre el diagnóstico de la HPo y la HPP era mayor que en los casos no operados25. En pacientes hospitalizados con HPo y CH, la HPP se detectó en el 2-16% de los casos25,26.
Con base en ligeras diferencias en cuanto a los criterios diagnósticos hemodinámicos, se reportan prevalencias de HPP del 5.3-8.5% en los candidatos a trasplante hepático ortotópico (THO)27,28. Lo relevante en este grupo es que el 65% de los casos con una presión sistólica de la arteria pulmonar (PSAP)>50mm Hg en el ecocardiograma transtorácico (ECOTT) tienen HPP corroborada hemodinámicamente29.
Un panorama obscuro de la historia naturalLa supervivencia (SV) media de los pacientes con HPP es de 15 meses. Sin embargo, existen diferencias dependiendo del tipo de HPo subyacente, pues aquellos pacientes del grupo de causa no hepática e HPP tienen una SV>90% a 5 años, mientras que en los casos con HPo hepática es del 68% a 5 años. Esto permite comprender que la mortalidad de los pacientes con HPP depende, principalmente, de la gravedad de la CH (mayor en Child-Pugh B/C), la hipertensión auricular derecha, un índice cardiaco bajo, una baja saturación venosa central y no recibir tratamiento como epoprostenol4,16. Así, las 2 principales causas de muerte en los pacientes con HPP son la descompensación de la falla cardiaca derecha en el 35% de los casos y las complicaciones relacionadas con la enfermedad hepática en otro 33%4.
La SV a 5 años de los pacientes con HPP es peor que en sus contrapartes con HAPI/HAP hereditaria (HAPH) (40 vs. 64%), a pesar de que los valores hemodinámicos sean mejores. Además, el número de hospitalizaciones en la HPP es mayor, y con más riesgo de fallecer después del primer internamiento (78 vs. 70%; p=0.004)20.
FisiopatologíaLos hallazgos histopatológicos son indistinguibles de otros subtipos de HAP, en los cuales las arterias pulmonares<500micras son afectadas con un espectro de lesiones obstructivas por todo el pulmón5,30.
Al igual que en los otros subtipos de HAP, no hay una explicación fisiopatológica satisfactoria, en parte debido a la falta de un modelo animal y a su baja prevalencia31, pero es claro que existe un estímulo detonante y una condición que lo perpetúa. Dentro de los detonantes se han postulado el estado hiperdinámico, el desequilibrio entre sustancias vasoconstrictoras, vasodilatadores y con efecto proliferativo celular en la circulación sanguínea (endotelina 1, interleucina 1, serotonina, glucagón, tromboxano B2, péptido vasoactivo intestinal e interleucina 6), y factores genéticos32,33. Incluso se ha planteado que en pacientes con CH la condición inflamatoria (endotoxemia, translocación bacteriana) puede participar en la remodelación vascular pulmonar34.
Las concentraciones de serotonina y endotelina 1 están aumentadas en los pacientes con CH e HPP (aumento en la síntesis esplácnica). Además, hay un predominio en la circulación pulmonar de receptores tipo A de endotelina 1 (tipo A: vasoconstricción; B: vasodilatación/degradación de endotelina 1). Otras alteraciones humorales son la disminución en la síntesis de prostaciclina (disminución de la sintetasa en arterias de pequeño y mediano calibre)35, y el aumento de la concentración sérica de interleucina 6 (remodelación vascular) con respecto a la interleucina 1 y el factor de necrosis tumoral tipo alfa36.
No se ha demostrado que exista relación con mutaciones de la proteína morfogenética ósea tipo 2 (BMPR2), ni de la cinasa similar al receptor de la activina (ALK-1) en los pacientes con HPP24, aunque existen polimorfismos de un solo nucleótido asociados al receptor tipo 1 de estrógeno, a la aromatasa31, a la fosfodiesterasa 5, la angiopoyetina 1 y la proteína de unión al calcio tipo 4. Se ha documentado además mutaciones en las vías del crecimiento celular/apoptosis (proteína S100A4, SERPINE1, RARB, CAV1, SMAD3, RUNX, RBPSUH)16,37.
El proceso diagnósticoTempranamente, hasta el 60% de los casos son asintomáticos38 o solo tienen síntomas y signos propios de la HPo. Pese a esto, la mayoría de los pacientes son diagnosticados en clase funcional (CF) iii-iv de la clasificación de la New York Heart Association4,17 y presentan manifestaciones características de la fase avanzada de la enfermedad, como disnea progresiva de esfuerzo (síntoma predominante de presentación), dolor torácico y/o síncope39. También pueden presentar signos de sobrecarga hídrica (ingurgitación yugular, hepatomegalia pulsátil, ascitis, edema de miembros pélvicos)39, que en el contexto de la HPo son difíciles de interpretar15. Es interesante que los pacientes con HPP no presentan habitualmente acropaquia, asterixis o edema de miembros pélvicos, además se asocian con una menor prevalencia de ascitis con respecto a aquellos con HPo sin HPP21.
En el registro REVEAL de los EUA, los pacientes con HPP tuvieron significativamente más fatiga (31 vs. 23%), edema (33 vs. 21%) y distensión abdominal (12 vs. 3%) que los pacientes con HAPI/HAPH. La frecuencia de presíncope/síncope fue la misma20.
En la exploración física del precordio los posibles hallazgos son: desplazamiento del choque de la punta hacia la línea axilar media, levantamiento sistólico paraesternal izquierdo bajo, palpación del cierre de la válvula pulmonar en el segundo espacio intercostal izquierdo (38%), auscultación de un desdoblamiento fijo del segundo ruido en el foco pulmonar (82%), además de un aumento en su intensidad de manera comparativa con el segundo ruido en el foco aórtico, un soplo sistólico en el foco tricuspídeo (61%) y un soplo diastólico en el foco pulmonar39.
También existe el grupo de pacientes con HPP evaluados para THO, los cuales pueden estar asintomáticos, y la única manera de sospechar la enfermedad es mediante los hallazgos del ECOTT, en especial el aumento en la presión sistólica del ventrículo derecho (PSVD)16.
Los parámetros de laboratorio no permiten distinguir plenamente a los pacientes con HPP de aquellos con HPo sin HP, aunque los niveles de transaminasas y de fosfatasa alcalina son menores, y la concentración de albúmina es mayor en los casos con HPP21. La concentración del fragmento N-terminal del propéptido natriurético cerebral y del péptido natriurético cerebral han mostrado su utilidad en los pacientes con HAPI40,41. En el registro REVEAL no se encontró diferencia en la concentración de ambos entre los pacientes con HAPI/HAPH e HPP20, aunque es útil para evaluar la respuesta al tratamiento en ambos grupos42.
Los pacientes pueden tener hipoxemia leve, hipocapnia y un aumento del gradiente alveoloarterial de oxígeno15. La combinación de una presión parcial arterial de bióxido de carbono<30mm Hg y un gradiente alveoloarterial de oxígeno >20mm Hg tiene un valor predictivo negativo del 90% para el diagnóstico de HPP a una altitud de 100m sobre el nivel del mar43,44. Sin embargo, no existe correlación entre los valores gasométricos y los parámetros hemodinámicos en esta población44. Las pruebas de funcionamiento respiratorio muestran un patrón restrictivo leve, con una disminución en la difusión de monóxido de carbono39. El electrocardiograma no se puede utilizar para diferenciar la HPP de otras formas de HP45, y la radiografía de tórax es normal en etapas tempranas, pero conforme progresa la enfermedad hay prominencia del tronco de la arteria pulmonar y de los hilios de manera bilateral, con una terminación abrupta de los vasos en la periferia y crecimiento del ventrículo derecho (VD)46. Los datos en la tomografía de tórax incluso contrastada son similares a los distintos subtipos de HAP (datos de sobrecarga de presión de las cavidades derechas, patrón de perfusión en mosaico, un diámetro del tronco de la arteria pulmonar >29mm, una relación>1 del diámetro de las arterias segmentarias y el bronquio correspondiente, y/o una relación>1 del diámetro del tronco de la arteria pulmonar y la aorta ascendente, especialmente en pacientes menores de 50 años)46. La gammagrafía ventilatoria/perfusoria pulmonar es normal o con defectos perfusorios difusos en parches47, sin que se identifiquen defectos segmentarios48. La prueba de caminata de 6minutos sirve para evaluar la CF de los pacientes con HPP49, y la distancia recorrida se relaciona con la CF y no con el índice de Child-Pugh4. Existe preocupación sobre la limitación de su utilidad en pacientes con ascitis y edema de tejidos blandos en las extremidades inferiores15, así como su falta de correlación con la SV de los pacientes4.
El ECOTT está indicado en los pacientes con HPo y síntomas sugestivos de HP, y en aquellos casos con HPo y CH evaluados para THO50. Aunque la mayoría de la literatura se enfoca en la medición de la PSVD y la PSAP, existen algoritmos de evaluación ecocardiográfica en pacientes con sospecha de HP, y en los que se resaltan otros parámetros de utilidad para el corazón derecho: velocidad telediastólica de regurgitación de la pulmonar, la relación del VD/ventrículo izquierdo, tiempo de aceleración del tracto de salida del VD, el desplazamiento del plano anular tricuspídeo en sístole, el tiempo de relajación isovolumétrica del VD, y el diámetro de la vena cava inferior con el grado de colapso inspiratorio. En caso de hallazgos anormales se deberán evaluar, además, el volumen de la aurícula derecha, índice de desempeño del VD, índices tisulares del VD, índice de excentricidad, gasto cardiaco, resistencia vascular pulmonar (RVP), datos de disfunción diastólica del ventrículo izquierdo, morfología/función valvular, y búsqueda de defectos cardiacos51.
La PSVD se calcula utilizando la velocidad pico de regurgitación tricuspídea (VTR) junto con la estimación de la presión de la aurícula derecha a través del grado de colapso inspiratorio de la vena cava inferior: PSVD=4(VTR)2+ presión de la aurícula derecha. Será importante descartar la presencia de un gradiente a través del tracto de salida del VD o de la válvula pulmonar (obstrucción) para poder considerar que la PSVD es igual a la PSAP52,53. Los resultados deben interpretarse con cuidado, pues hasta un 20% de los pacientes candidatos a THO tienen aumentos moderados de la presión pulmonar no debida a HPP. Además, a pesar de que existen datos que señalan que el ECOTT tiene una sensibilidad del 97% y una especificidad del 77% para detectar una PMAP≥35mm Hg por cateterismo cardiaco derecho (CTTD) en HPP53, también es importante señalar que, en un estudio, donde el 89% de los sujetos tenían HP por cateterismo cardiaco, hasta en el 50% de los casos la PSAP obtenida invasivamente tenía una diferencia≥10mm Hg con respecto al ECOTT, y el problema radicaba, primordialmente, en la presión de la aurícula derecha estimada51,54.
Con lo mencionado previamente, no está bien definido el punto de corte de PSVD/PSAP en el ECOTT para recomendar la evaluación hemodinámica de los pacientes candidatos a THO. En un estudio se concluyó que una PSAP>38mm Hg detectada por ECOTT es bastante sensible, específica y precisa, e incluso su especificidad se incrementa un poco si al valor de la PSAP se suma la dilatación del VD (diámetro telediastólico≥3.3cm)55.
Actualmente, se recomienda clasificar por los hallazgos ecocardiográficos a los pacientes con sospecha de HP en: 1) baja probabilidad para HP (VTR≤2.8cm/s o no medible sin otros datos de HP); 2) probabilidad intermedia para HP (unVTR≤2.8cm/s o no medible con otros datos de HP, o un VTR de 2.9-3.4cm/s en el ECOTT), o 3) probabilidad alta para HP (unVTR de 2.9-3.4cm/s con otros datos de HP o un VTR>3.4cm/s). Un paciente con HPo y sospecha de HP en el primer grupo solo requiere seguimiento ecocardiográfico (iia/C), pues no se puede descartar el desarrollo de HPP en períodos cortos de 2 a 3 meses13,28,55. En el segundo grupo se puede considerar el CTTD (iia/B), mientras que en el tercero se debe someter el paciente a CTTD (I/C)13. Por otro lado, están los pacientes con HPo evaluados para THO, y en los cuales no hay un corte bien definido de PSVD/PSAP para referir al paciente a CTTD. Existen algunos grupos que hacen sugerencias al respecto: 1) todos los pacientes candidatos a THO con una PSAP≥45mm Hg (Asociación Americana para el Estudio de las Enfermedades Hepáticas o AASLD)56; 2) el algoritmo de la Clínica Mayo en Rochester, Minnesota, EUA utiliza una PSVD>50mm Hg27; y 3) se pudiera considerar en aquellos casos con PSVD de 30-50mm Hg con dilatación y/o disfunción del VD por ECOTT57. La Asociación Europea para el Estudio del Hígado (EASL) no hace ninguna recomendación al respecto58.
Las mediciones en el CTTD permiten identificar a los pacientes del grupo 2 de HP y a aquellos con estado hiperdinámico45. El diagnóstico hemodinámico de la HPP es similar al de la HAP, es decir HP precapilar (tabla 2), pero con algunas particularidades (tabla 3). Aunque no se requiere para la definición actual de HAP, se recomienda conocer el valor de RVP. El valor normal es hasta 2 unidades Wood (UW), pero emplear un corte de 3UW o 240dyn/s/cm−5 se justifica por el hecho de que la probabilidad de HAP con RVP<3UW es baja59. Para el caso de la HPP es un requisito que la RVP sea>3UW (>240dyn/s/cm−5) para el diagnóstico45.
Los pacientes con HPo con PMAP≥25mm Hg y presión de oclusión de la arteria pulmonar (POAP) o presión en cuña de la arteria pulmonar (PCAP)>15mm Hg, cumplen con la definición hemodinámica de HP poscapilar, sin embargo, la presencia de RVP>3UW orientan a la coexistencia de una HP precapilar13. En la serie de la Clínica Mayo, este grupo correspondió al 24% de los pacientes con RVP>3UW. Lo más importante es que el 100% de estos casos tuvieron un gradiente transpulmonar de presión (GTP) (GTP=PMAP−POAP/PCAP)>12mm Hg (vs. 27% en los que tuvieron una RVP<3UW). Otro dato relevante es que este subgrupo de HPP no tiene mayor grado de disfunción diastólica del ventrículo izquierdo, ni dilatación de la aurícula izquierda por el ECOTT29, lo que los diferenciaría de aquellos pacientes del grupo 2 de la OMS para HP.
Un dato interesante es que el GTP se ve influenciado por el flujo sanguíneo, la resistencia al flujo y las presiones de llenado de las cavidades izquierdas (determinantes de la PMAP), mientras que el gradiente diastólico de presión (GDP) (GDP=presión diastólica de la arteria pulmonar−POAP/PCAP) no está influenciado por los determinantes de la PMAP, ni de la POAP/PCAP, lo que parece ser una desventaja del GTP. Y aunque no se incluyeron pacientes con HPP cabe señalar que durante un estudio se encontró que en aquellos casos con HP poscapilar, un GTP>12mm Hg y un GDP≥7mm Hg, la SV fue similar a la HAPI60. Hasta la fecha se recomienda el uso del GTP para el diagnóstico de HPP en el escenario de HP poscapilar.
La prueba de vasodilatación pulmonar aguda durante el CTTD (óxido nítrico inhalado, adenosina iv y epoprostenol iv), solo se recomienda en los pacientes con HAPI, HAPH y HAP inducida por fármacos/toxinas13. En la HPP hay un 0.7-1.5% de respondedores a la prueba de vasodilatación pulmonar aguda (disminución≥10mm Hg de la PMAP con un valor final de la misma de≤40mm Hg)17,61, aunque la tasa de respuesta persistente a los calcioantagonistas orales es del 0%61. Además, los calcioantagonistas orales producen un incremento del GPVH debido a un aumento en el gasto cardiaco, lo que limita su uso de manera crónica62–64. Durante un estudio el uso de 5-mononitrato de isosorbide oral durante la prueba de vasodilatación pulmonar aguda produjo mejor respuesta vasodilatadora que el óxido nítrico inhalado y el epoprostenol iv65, aunque su uso no es regular.
No se recomienda la medición de rutina del GPVH (GPVH= presión venosa hepática libre–presión venosa hepática en cuña) durante el CTTD, pero es recomendable para confirmar el diagnóstico de HPo. Se debe de medir la presión venosa hepática libre en el interior de una vena suprahepática, 2-4cm de la desembocadura de la misma en la vena cava inferior, siendo importante que la diferencia de presión no sea mayor a 2mm Hg con respecto a la vena cava inferior. Posteriormente, se infla el globo del catéter, se corrobora la oclusión total mediante la inyección de 5ml de contraste y se espera 60 o más segundos para obtener la presión venosa hepática en cuña48,66. En pacientes con CH descompensada con ascitis refractaria, el GPVH en los casos con HPP fue de 35.2±2.5mm Hg (presión portal por punción 34.7±1.8mm Hg)26, mientras que en otro estudio los pacientes con HPo que fueron sometidos a biopsia hepática transyugular con una PMAP≥25mm Hg, POAP≤13mm Hg y RVP>3UW (15 sujetos) tuvieron un mediana del GPVH de 15(10)mm Hg25.
Es importante tener en cuenta que las condiciones hemodinámicas pulmonares pueden cambiar con el tiempo en los casos con HPP. Así, es bastante recomendable la evaluación con CTTD cada 12 meses en los candidatos a THO con HPP27. Aunque esto dependerá de las políticas de cada uno de los centros.
Los diferentes pasos en el tratamientoAl igual que el resto de los pacientes con HAP el manejo consiste en medidas generales, terapia de soporte y terapia específica.
Medidas generalesSe debe evitar el embarazo. Se recomienda la inmunización anual para influenza y contra neumococo conforme a los lineamientos nacionales. Es importante el soporte psicosocial por el impacto que la enfermedad tiene sobre la calidad de vida de los pacientes. Además, se sugiere el uso de oxígeno en los vuelos en avión en pacientes en CF III-IV de la OMS y con una presión parcial de oxígeno<60mm Hg. En caso de ser necesaria una cirugía preferentemente deberá de emplearse anestesia epidural (difícil en el contexto de trombocitopenia). Y se debe de evitar la actividad física excesiva porque puede producir síntomas de bajo gasto cardiaco13.
Terapia de soporteEl registro COMPERA, incluyó a 60 pacientes con HPP (el 30% anticoagulados), y no mostró que la anticoagulación oral brindara un beneficio en cuanto a la SV a 3 años en HAP no idiopática67.
Los pacientes con HPP e HPo hepática pueden presentar edema de tejidos blandos, así como ascitis. El manejo habitual en la HPo hepática es la combinación de furosemida con espironolactona en una relación de 40 y 100mg, respectivamente (máximo 160mg/400mg)68. Así, aunque no existen estudios aleatorizados en pacientes con HAP, el uso de furosemida mejora la sintomatología y previene la descompensación de la falla cardiaca13. Además, existe evidencia del papel que la aldosterona puede desempeñar en la fisiopatología de la HAP, lo cual puede ser el sustento de que la combinación de espironolactona con ambrisentan, un antagonista del receptor de endotelina (ARE), tiene mejores resultados que el ARE aislado69. Es importante resaltar que en el contexto de la HPP el uso de diuréticos puede inducir encefalopatía hepática, lesión renal aguda, ginecomastia, desórdenes de electrólitos y calambres, principalmente, en las primeras semanas de uso. La frecuencia de estas complicaciones se puede disminuir utilizando como metas la disminución de 0,5kg de peso al día si solo hay ascitis clínicamente demostrable, y hasta 1kg si hay ascitis con edema periférico, disminuyendo la dosis una vez que no haya evidencia clínica de ascitis70.
En este escenario, el uso de bloqueadores de canales de calcio no ha demostrado respuesta y se debe recordar que pueden producir incremento en la retención de líquidos, pueden reducir la función ventricular derecha e incrementar la presión portal por lo que se deben evitar en HPP71.
El oxígeno suplementario de manera ambulatoria solo debe prescribirse si la presión parcial de oxígeno<60mm Hg con evidencia de mejoría sintomática y de la oxigenación con su uso13.
Hay 2 fármacos cardiovasculares de interés: 1) la digoxina de la cual no se sabe su papel con el uso crónico en los pacientes con HAP13, y 2) los betabloqueadores, que se utilizan para la profilaxis del sangrado variceal, sin embargo, se ha demostrado que en casos con HPP moderada a grave su uso se asocia con una disminución de la distancia y mayor grado de disnea en la prueba de caminata de 6minutos, y caída del gasto cardiaco en el CTTD72. Se deberá considerar evitar los betabloqueadores si es totalmente posible73 y, preferentemente, utilizar terapias endoscópicas si está indicado71,74.
La deficiencia de hierro es frecuente en la HAP, aunque no hay estudios controlados de los efectos de la terapia de sustitución13. En los casos con HPo existe el riesgo inherente de pérdidas crónicas de sangre a través del tubo digestivo75, y aunque es recomendable la sustitución con hierro no se sabe los efectos a largo plazo en el hígado, ni la meta de hemoglobina en este contexto.
Terapia específicaEn este apartado quedan incluidos los inhibidores de la fosfodiesterasa 5, los análogos de prostaciclina o prostanoides intravenosos, subcutáneos, inhalados u orales, los ARE, y los estimulantes de la guanilato ciclasa soluble.
Los pacientes con HPP han sido excluidos de casi todos los estudios aleatorizados y controlados, excepto del estudio PATENT (riociguat), que incluyó a 13 pacientes (2 con placebo y 11 con riociguat 2.5mg 3 veces al día), aunque no se hizo ningún análisis de los subgrupos13,45,76. A pesar de esto existen múltiples estudios no aleatorizados que incluyen alrededor de 400 pacientes con HPP, y que muestran que la SV a 5 años de aquellos que recibieron alguna terapia específica fue del 40-68%, mientras que sin esta solo fue del 2-14%77.
El registro REVEAL mostró diferencias en cuanto al uso de terapia específica en la HPP. Así, los casos diagnosticados previamente recibían vasodilatadores específicos más frecuentemente que los de diagnóstico reciente (84 vs. 57%). A pesar de esto, la frecuencia de administración de estos fármacos en los casos previamente diagnosticados con HAPI/HAPH fue mayor que en sus contrapartes con HPP, lo que no se observó en los casos de reciente diagnóstico de ambos grupos de HAP. Por último, el grupo de terapia específica más utilizado en la HPP a los 3 y 12 meses de inclusión en el registro fue el de los inhibidores de la fosfodiesterasa 5, seguidos por los análogos de prostaciclina intravenosos/subcutáneos, los análogos de prostaciclina inhalados/orales y el grupo menos empleado fue el de los ARE20. La limitación en el uso de los ARE radica, principalmente, en el riesgo de hepatotoxicidad, sin embargo, la evidencia muestra que el perfil de seguridad en la HPP con CH es igual al de los pacientes con HAPI sin CH. La única recomendación es el seguimiento mensual de las pruebas de funcionamiento hepático45.
Con base en la historia natural y lo expuesto en los párrafos anteriores existe un consenso para utilizar la terapia específica en los pacientes con HPP. En cuanto a esto se pueden identificar 2 escenarios, el primero en el contexto de los pacientes candidatos a THO con HPP moderada a grave27, y el segundo en aquellos casos no candidatos a THO, pero que se encuentran por lo menos en CF II de la OMS13.
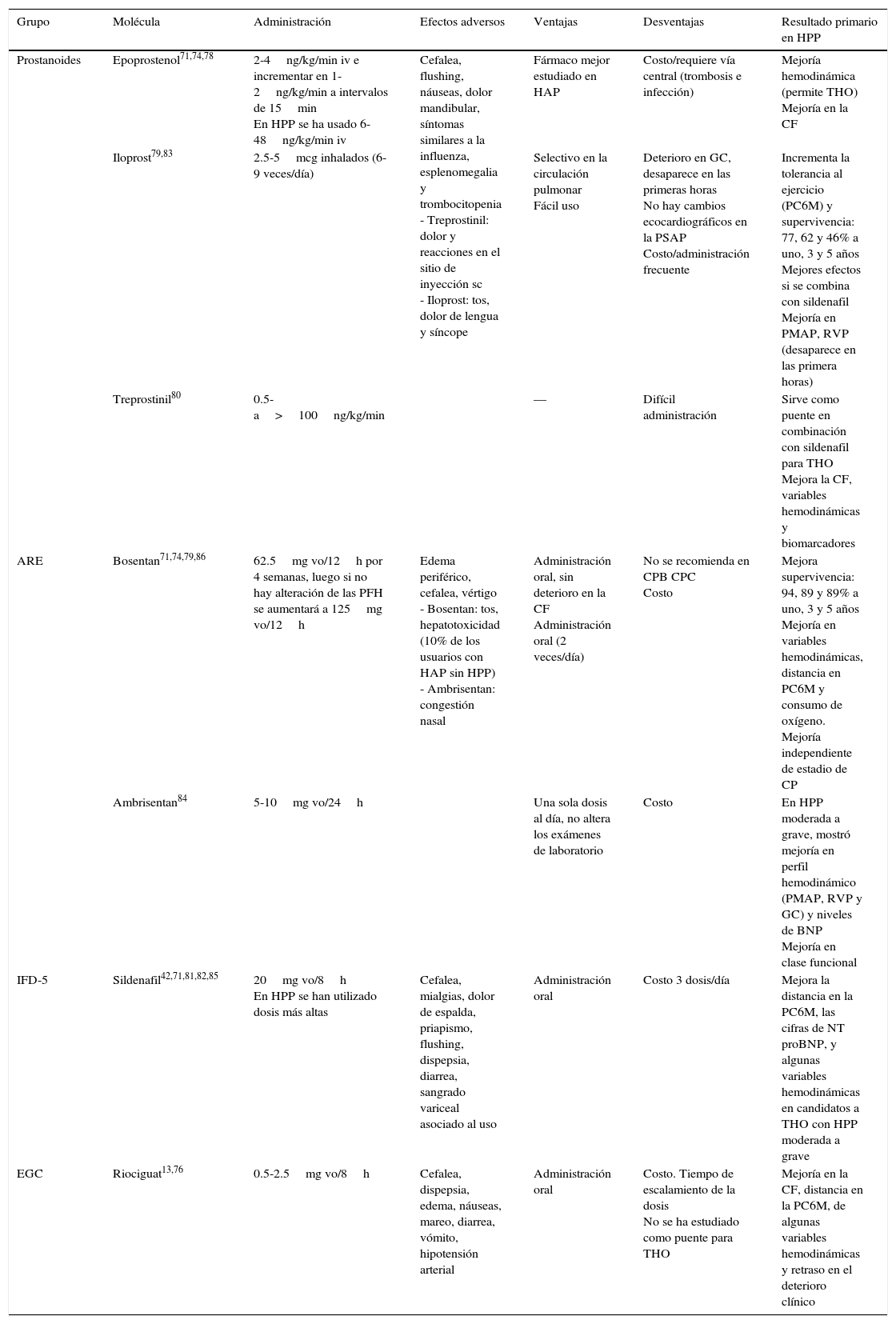
En la tabla 4 se exponen los diferentes vasodilatadores pulmonares de los que se tiene conocimiento de su uso en casos con HPP13,42,74,76,78–86.
Terapia específica en pacientes con hipertensión portopulmonar
| Grupo | Molécula | Administración | Efectos adversos | Ventajas | Desventajas | Resultado primario en HPP |
|---|---|---|---|---|---|---|
| Prostanoides | Epoprostenol71,74,78 | 2-4ng/kg/min iv e incrementar en 1-2ng/kg/min a intervalos de 15min En HPP se ha usado 6-48ng/kg/min iv | Cefalea, flushing, náuseas, dolor mandibular, síntomas similares a la influenza, esplenomegalia y trombocitopenia - Treprostinil: dolor y reacciones en el sitio de inyección sc - Iloprost: tos, dolor de lengua y síncope | Fármaco mejor estudiado en HAP | Costo/requiere vía central (trombosis e infección) | Mejoría hemodinámica (permite THO) Mejoría en la CF |
| Iloprost79,83 | 2.5-5mcg inhalados (6-9 veces/día) | Selectivo en la circulación pulmonar Fácil uso | Deterioro en GC, desaparece en las primeras horas No hay cambios ecocardiográficos en la PSAP Costo/administración frecuente | Incrementa la tolerancia al ejercicio (PC6M) y supervivencia: 77, 62 y 46% a uno, 3 y 5 años Mejores efectos si se combina con sildenafil Mejoría en PMAP, RVP (desaparece en las primera horas) | ||
| Treprostinil80 | 0.5-a>100ng/kg/min | — | Difícil administración | Sirve como puente en combinación con sildenafil para THO Mejora la CF, variables hemodinámicas y biomarcadores | ||
| ARE | Bosentan71,74,79,86 | 62.5mg vo/12h por 4 semanas, luego si no hay alteración de las PFH se aumentará a 125mg vo/12h | Edema periférico, cefalea, vértigo - Bosentan: tos, hepatotoxicidad (10% de los usuarios con HAP sin HPP) - Ambrisentan: congestión nasal | Administración oral, sin deterioro en la CF Administración oral (2 veces/día) | No se recomienda en CPB CPC Costo | Mejora supervivencia: 94, 89 y 89% a uno, 3 y 5 años Mejoría en variables hemodinámicas, distancia en PC6M y consumo de oxígeno. Mejoría independiente de estadio de CP |
| Ambrisentan84 | 5-10mg vo/24h | Una sola dosis al día, no altera los exámenes de laboratorio | Costo | En HPP moderada a grave, mostró mejoría en perfil hemodinámico (PMAP, RVP y GC) y niveles de BNP Mejoría en clase funcional | ||
| IFD-5 | Sildenafil42,71,81,82,85 | 20mg vo/8h En HPP se han utilizado dosis más altas | Cefalea, mialgias, dolor de espalda, priapismo, flushing, dispepsia, diarrea, sangrado variceal asociado al uso | Administración oral | Costo 3 dosis/día | Mejora la distancia en la PC6M, las cifras de NT proBNP, y algunas variables hemodinámicas en candidatos a THO con HPP moderada a grave |
| EGC | Riociguat13,76 | 0.5-2.5mg vo/8h | Cefalea, dispepsia, edema, náuseas, mareo, diarrea, vómito, hipotensión arterial | Administración oral | Costo. Tiempo de escalamiento de la dosis No se ha estudiado como puente para THO | Mejoría en la CF, distancia en la PC6M, de algunas variables hemodinámicas y retraso en el deterioro clínico |
ARE: antagonista del receptor de endotelina; BNP: péptido natriurético cerebral; CF: clase funcional; CP: Child-Pugh; CPB: Child-Pugh B; CPC: Child-Pugh C; GC: gasto cardiaco; HAP: hipertensión arterial pulmonar; HPP: hipertensión portopulmonar; IFD-5: inhibidores de la fosfodiesterasa 5; iv: intravenoso; NT-proBNP: fragmento N terminal del péptido natriurético cerebral; PC6M: prueba de caminata de 6minutos; PFH: pruebas de funcionamiento hepático; PMAP: presión media de arteria pulmonar; PSAP: presión sistólica de la arteria pulmonar; RVP: resistencia vascular pulmonar; sc: subcutánea; THO: trasplante hepático ortotópico; vo: vía oral.
Es interesante que la respuesta a la terapia específica, aparentemente, pudiera verse comprometida por la presencia de cortocircuitos portosistémicos espontáneos grandes (>10mm) en pacientes con CH (modelo para la enfermedad hepática avanzada [MELD]±12) y HPP moderada y grave. Su presencia es frecuente (71-87%), y aunque no parecen estar relacionados con el desarrollo de HPP (presencia en sujetos con CH sin HP), no es claro su papel hasta ahora87.
El seguimientoEn los pacientes con HAP en tratamiento con vasodilatadores específicos el seguimiento se ajustará a las características del sujeto y dependerá del centro, es decir que puede ser solo con revisiones clínicas, exámenes de laboratorios y ECOTT, o incluso con CTTD de manera regular13. En el caso de los pacientes con HPP se recomienda la evaluación de la respuesta a la terapia específica cada 3 meses57. Los resultados de la prueba de caminata de 6minutos durante el seguimiento en este grupo pueden verse afectados por manifestaciones clínicas propias de la CH15.
El impacto del trasplante hepáticoA pesar de que el 90% de los casos de HPP en Occidente están relacionados con CH12,13, la indicación de THO dependerá de la gravedad de la enfermedad hepática/HPo (tabla 5)27,56. A diferencia del síndrome hepatopulmonar, la HPP como una «indicación» de THO es controversial, pues la respuesta hemodinámica postrasplante no es predecible88. La mayoría de los pacientes con HPP pueden ser retirados de terapia vasodilatadora intravenosa luego del THO, y hasta la mitad pueden ser retirados de la terapia oral, sin embargo, no se sabe con claridad qué pacientes se pueden retirar exitosamente del tratamiento89. Además, hay que recordar que los candidatos a THO con HPP tienen 2.46 veces más riesgo de muerte que aquellos sin HPP83, y eso no es todo, ya que los casos que tienen una PMAP>45mm Hg, o>35mm Hg y/o RVP>400dyn/s/cm−5 (5UW) tienen una mortalidad perioperatoria del 50-100%90.
Indicaciones para trasplante hepático ortotópico y combinado
| Hepático27,79 | Descompensación de la cirrosis hepática: ascitis, encefalopatía hepática, sangrado variceal; o Un puntaje del modelo para la enfermedad hepática avanzada o MELD≥15 |
| Combinado | |
| Pulmón-hígado84 | Enfermedad vascular pulmonar con: |
| a. CF III o IV de la New York Heart Association a pesar de por lo menos 3 meses de terapia combinada incluidos prostanoides | |
| b. Índice cardiaco<2l/min/m2 | |
| c. PAD>15mm Hg | |
| d.<350m (PC6M) | |
| e. Desarrollo de hemoptisis o derrame pericárdico significativos, o datos de falla cardiaca derecha progresiva (insuficiencia renal, hiperbilirrubinemia, aumento del BNP o ascitis recurrente) | |
| Con evidencia histológica de cirrosis más un GPVH>10mm Hg | |
| a. Evitar si: albúmina<2g/dl, INR>1.8 o en presencia de ascitis o encefalopatía grave | |
| Pulmón-corazón-hígado84 | Arriba mencionado más: |
| a. Disfunción miocárdica o defectos, o defectos de las cavidades cardiacas irreparables | |
BNP: péptido natriurético cerebral; CF: clase funcional; GPVH: gradiente de presión venosa hepática; INR: cociente normalizado internacional; MELD: modelo para la enfermedad hepática avanzada; PAD: presión de la aurícula derecha; PC6M: prueba de caminata de 6 minutos.
La terapia específica desempeña un papel importante para el THO. Ashfaq et al. encontraron que los pacientes con HPP moderada a grave que recibieron terapia con vasodilatadores específicos (16 pacientes) tuvieron una SV del 75, 69 y 43% a uno, 2 y 5 años, respectivamente. Mientras que, de este grupo, aquellos que respondieron a la terapia y fueron trasplantados (11 casos) tuvieron una SV>70% a partir del primer año de seguimiento84. Swanson et al. encontraron que en los pacientes con HPP tratados y sometidos a THO (9 casos) la SV fue>70% a un año y>65% hasta los 9 años de seguimiento91. Khaderi et al. en 7 pacientes con HPP tratados y con THO tuvieron una SV del 86% a 7.8 años85.
Actualmente, se recomienda que los sujetos con HPP leve pueden recibir un THO sin necesidad de terapia específica, mientras que en el resto de los casos se ha propuesto utilizar la excepción del MELD de los EUA27,71,83,89,90.
- 1)
HPP moderada a grave (diagnóstico hemodinámico):
- a.
PMAP≥35mm Hg, RVP≥240dyn/s/cm−5 (3UW), y POAP/PCAP≤15mm Hg.
- a.
- 2)
Mejoría documentada con la terapia específica vasomoduladora (mínimo 12 semanas):
- a.
PMAP<35mm Hg, RVP<400dyn/s/cm−5 (5UW), y función satisfactoria del VD por ECOTT (definición no estandarizada que depende del centro).
- a.
- 3)
Se deberá actualizar cada 3 meses la excepción de MELD si se cumplió el criterio hemodinámico de respuesta (punto 2).
- a.
Agregar 2 puntos del puntaje de MELD
- a.
La Red de Procuración y Trasplante de Órganos (EUA) recomienda que a estos pacientes se les asigne 22 puntos de MELD de manera inicial71,92 para acortar los tiempos de espera para un THO. A pesar de las recomendaciones, menos de la mitad de los pacientes con excepción de MELD y HPP en la lista de espera en los EUA cumplen estos criterios estandarizados83.
No se puede concluir nada acerca de la utilidad del trasplante combinado pulmón-hígado o pulmón-corazón-hígado en pacientes con HPP (tabla 5), pues hasta el 2014 se habían realizado 58 trasplantes de este tipo por diversas indicaciones (SV de 20 a 43% a 1 año)93,94, y hasta el 2011 solo 10 en HPP (6 trasplantes combinados pulmón-hígado y 4 trasplantes combinados pulmón-corazón-hígado) con 2 fallecimientos en las primeras 24h (choque cardiogénico) y SV de 2-140 meses en el resto86.
En algunos casos existe una normalización hemodinámica postrasplante, sin embargo, se desconoce si realmente refleja la curación de la enfermedad vascular pulmonar88. A pesar del optimismo en algunos grupos, el porcentaje de pacientes que permanecen con terapia específica es variable (18-50%), no se sabe la razón, pero pudiera estar relacionada con la etiología de la HPo (mejor respuesta en enfermedad hepática asociada a ingesta de alcohol) y/o a la definición de «mejoría» hemodinámica postrasplante85.
Finalmente, la HPP es un reto para el anestesiólogo, pues aumenta el riesgo de complicaciones trans- y postoperatorias en el THO. Los cambios mayores en la hemodinámica pulmonar con la repercusión se consideran un reto clínico71. Los momentos críticos son durante la inducción anestésica (disminución del tono vasomotor, presión positiva de la ventilación mecánica invasiva), la fase preanhepática (infusión de cristaloides, coloides y productos sanguíneos), la fase anhepática con el pinzamiento de la vena cava, vena porta y arteria hepática (caída del gasto cardiaco del 40-50%), y la reperfusión del órgano (aumento en la PMAP asociado a sustancias vasoactivas e incremento del gasto cardiaco)34,89. Así, junto con una cirugía>12h, la presencia de una presión arterial media<40mm Hg (un episodio de≥2mins) y una PMAP>40mm Hg (3 episodios de≥2min separados entre sí) durante el transoperatorio, se asocian con pobre funcionamiento del injerto tempranamente, disfunción primaria del injerto o muerte por causas hemodinámicas (infarto del miocardio o arritmias cardiacas)95. En ocasiones es necesario que se empleen algunos fármacos cardiovasculares como inotrópicos, vasopresores e incluso un sistema de circulación extracorpórea venovenoso34,89. De manera inicial en la fase postoperatoria se debe ser restrictivo con el manejo de líquidos intravenosos en este grupo de pacientes, y en caso de haber deterioro de la función renal se debe considerar la hemodiálisis venovenosa continua96. Además, hay que recordar que la respuesta hemodinámica pulmonar postrasplante es impredecible, así que se debe de tener en consideración la posibilidad de utilizar terapias específicas. Aunque no existen estudios aleatorizados para el manejo de la HP en el postoperatorio, junto con las medidas previamente mencionadas se puede emplear para el control de la PMAP y RVP prostanoides (epoprostenol iv e iloprost inhalado), óxido nítrico inhalado (costoso, con pobre respuesta en los pacientes con HP «fija», riesgo de síndrome de supresión –hipotensión arterial y deterioro de la oxigenación–), y sildenafil (respuesta equiparable al epoprostenol iv). Asimismo se ha planteado la posibilidad de utilizar estos fármacos en combinación en búsqueda de un efecto aditivo (epoprostenol iv+óxido nítrico inhalado, iloprost inhalado+óxido nítrico inhalado, sildenafil+óxido nítrico inhalado para evitar el síndrome de supresión). Por último, se deberá de mantener medidas de ventilación mecánica invasiva con metas de protección pulmonar y optimizar la oxigenación97.
ConclusionesLa frecuencia de la HPP en México se desconoce, y su estudio se encuentra en una etapa inicial. Probablemente, en cuanto el número de THO aumente en nuestro país se tendrán cifras aproximadas de la prevalencia de este padecimiento. El diagnóstico, finalmente, debe de ser hemodinámico, y durante la valoración diagnóstica en el escenario de sospecha de HP, así como en los candidatos a THO que cumplan con el criterio ecocardiográfico utilizado por el centro hospitalario para la evaluación hemodinámica. El uso de las terapias específicas parece tener un papel en el manejo de cierto grupo de pacientes candidatos a THO con HPP, así como en el tratamiento trans- y postoperatorio. En México existen algunos centros hospitalarios con unidades especializadas en el diagnóstico y tratamiento de la enfermedad vascular pulmonar. Habrá que crear un enlace entre estas y los centros con alto volumen de pacientes con CH, además con los equipos de THO que en el país existan, teniendo en cuenta que no es necesario que cada centro de THO cuente con una unidad de enfermedad vascular pulmonar, sino que se haga efectiva y ágil la mecánica de referencia.
FinanciaciónNinguna.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.