




The term autoimmune enteropathy (AIE) was applied to a form of “intractable diarrhoea” with serum gut autoantibodies, characterized by male predominance, early onset, poor response to parenteral nutrition and several autoimmune diseases, mainly type 1 diabetes. In recent years the vague concept of AIE has became more precise thanks to the discovery of its genetic and molecular basis. The FOXP3 molecule is crucial for the generation and maturation of regulatory T cells (Treg) expressing CD4+ and CD25+ molecules. Mutations of the FOXP3 gene, located in X chromosome, produce a syndrome with Immune dysfunction, Polyendocrinopathy, Enteropathy and X-linked inheritance (IPEX). The majority of the ancient so-called AIE cases probably correspond to the new IPEX syndrome, even in female patients who may have some autosomal genetic variants. Besides FOXP3, other molecules are likely to be involved in the generation and function of Treg and its deficiency may also enhance autoimmune disease and IPEX-like syndromes. Meanwhile, the important pathogenic role previously ascribed to gut autoantibodies has vanished, with it remaining as having only certain screening usefulness.
Avery proposed the term “Intractable diarrhoea” in 1968, to describe patients with unremitting chronic diarrhoea occurring in infants less than 3 months of age with the result of progressive malnutrition, after excluding an infectious aetiology.1 In spite of using intravenous feeding, mortality was frequent and occurred too early, before a complete study could be performed. This term may include a heterogeneous group of diseases (Table 1).
Different forms of intractable diarrhoea of infancy
| 1. Congenital diarrhoea |
| Syndromic or phenotypic diarrhoea |
| Microvillous inclusion disease |
| Tufting enteropathy |
| Congenital diarrhoea with mutant neurogenin-3 |
| Congenital diarrhoea with disorder of glycosylation |
| 2. IPEX and IPEX-like syndromes |
| 3. Other autoimmune enteropathies |
The very rare cases described as “Congenital diarrhoea” may be considered as intractable diarrhoea, however a group with early but no neonatal onset was separated from the congenital diarrhoea group. Later, when intestinal biopsies were easily available, a villous atrophy was frequently recorded, usually severe. Crypts can be hypoplastic, with few mitoses, resembling a graft versus-host disease, or hyperplastic, resulting in a picture indistinguishable from a celiac disease. An inflammatory infiltrate of the lamina propria is always present, thought it is not clear whether gut autoantibodies precede or follow intestinal inflammation.2
Autoimmune enteropathyWalker-Smith,3 Unsworth,4 and later, Savage5 investigated autoantibodies in patients with non-congenital intractable diarrhoea that showed villous atrophy, complement fixing gut autoantibodies, and associated to some cases, diabetes mellitus or hypothyroidism.
AIE is a rare familial disorder characterized by protracted diarrhoea, lack or poor response to parenteral nutrition, circulating antibodies and/or autoimmune diseases in infants without severe immunodeficiency. It is characterized by a somewhat more delayed onset than congenital enteropathies, often at 4–6 months of age and frequently has other evidence of autoimmune diseases. Dysmorphic features are common in congenital enteropathies but not in AIE.6
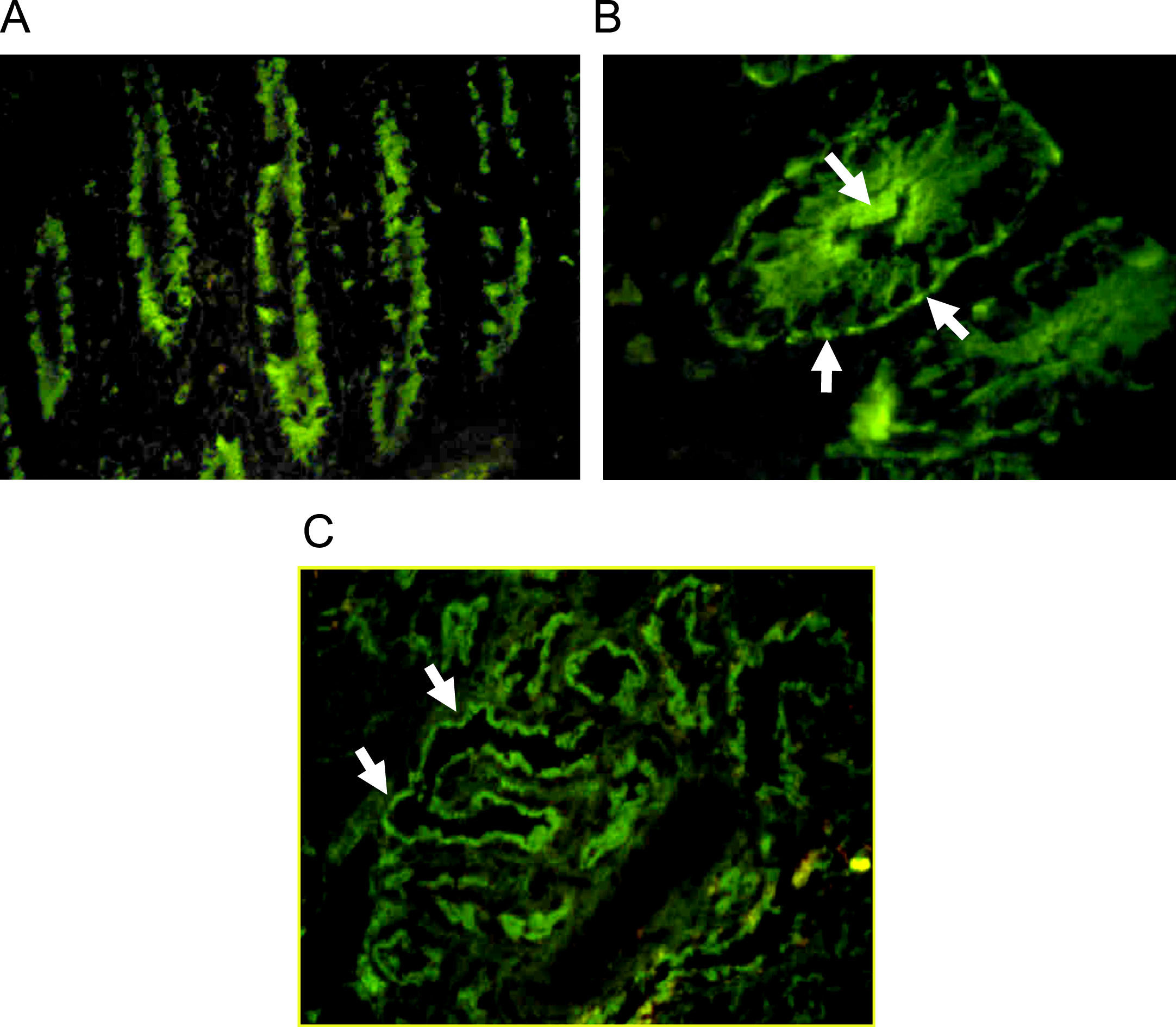
Autoantibodies were usually determined on human blood group 0 duodenal samples obtained from patients undergoing surgical procedures, although tissue from monkey could be used with satisfactory sensibility.7 A cross reaction between gut autoantibodies and renal epithelial cells is commonly reported.8 Immunofluorescence analysis showed two different patterns: complement-fixing apical antibodies and cytoplasmatic positive staining (Fig. 1). According to the report of Martin-Villa et al.9 patients with complement-fixing apical antibodies may have abnormal T-cell responses in vitro, and a cytoplasmatic pattern was associated to immunoglobulin deficiency, suggesting that different cellular or humoral immunologic alterations may favour a different gut autoantibody pattern. Although a correlation between serum autoantibodies levels and disease activity was found in some cases,8 the pathogenic role of gut autoantibodies has never been proven.
, the staining is stronger at brush-border of crypts. B. IgG positive antibodies on a cross section of gut (×100), the immunofluorescence is cytoplasmatic with more intensity over apical border and a think linear staining on basolateral border. C. Staining on monkey kidney showing IgG autoantibodies that react with tubular border but avoiding the glomerulus (×40).")
Indirect immunofluorescence pattern putting the patient's serum on frozen monkey tissue. A. IgA positive antibodies on a longitudinal section of gut (×40), the staining is stronger at brush-border of crypts. B. IgG positive antibodies on a cross section of gut (×100), the immunofluorescence is cytoplasmatic with more intensity over apical border and a think linear staining on basolateral border. C. Staining on monkey kidney showing IgG autoantibodies that react with tubular border but avoiding the glomerulus (×40).
High expression of HLA-A, -B, and -C with a stronger reactivity is commonly found in the crypt epithelium. An aberrant expression of DR molecules on the gut surface and crypt epithelium was observed, with a high number of DR positive activated cells in the lamina propria. Nevertheless the expression of HLA-DP and DQ was normal. The inappropriate DR expression in the epithelium is not an exclusive sign of autoimmunity, accompanying also chronic inflammatory processes, such as Crohn disease.
Several authors investigated the nature of the gut antigen in AIE. Coletti, et al.8 in 1991, using Western immunoblot, identified a 55kD protein located in gut and renal epithelial cells that reacted with serum autoantibodies. Later, Kobayashi10 detected a 75kD autoantigen, which was distributed through the whole intestine and kidney, and likely implicated in the development of nephropathy, although not present in the thyroid gland. A tryptophan hydroxylase (TH) was identified as the intestinal autoantigen in autoimmune polyendocrine syndrome type 1 which is accompanied by gastrointestinal symptoms in 20–30% of the cases.11 This antigen is mainly present in enterochromaffin cells of the mucosa, and the search for autoantibodies to TH is also recommended in patients with Crohn disease or ulcerative colitis.
The majority of reported patients with AIE were young boys with the onset of clinical manifestations at a very early stage, within the first 6 months of life, although it has also been observed in older children and female subjects, interestingly with less bowel involvement.12 Several reports describe the involvement of both the small and large bowels. Whether either regions of the gut are simultaneously affected or one region is affected before the other is unclear.13
It was presumed that extra-intestinal involvement was commonly present, but in the absence of a systemic evaluation some histological lesions outside gastrointestinal tract could remain unnoticed. Pancreatic lesions, with endocrine or exocrine insufficiency were usually observed, and diabetes mellitus was frequently the first manifestation of the disease, prior to diarrhoea. Atopic eczema showed the typical clinical and histological findings. During remissions the eczema improves and serum IgE levels decreased. Renal involvement is common and biopsy may show interstitial inflammatory nephritis or membranous glomerulonephritis.8,14 The liver injury was usually revealed by hepatomegaly and a moderate increase in serum transaminases. Histology shows a periportal fibrosis, which could be more extensive in some cases, associated to a clinical picture of chronic aggressive hepatitis. Thyroid insufficiency was noted in some cases, with lymphoid cell infiltration that could be accompanied by fibrosis.
The mortality of patients with AIE was very high, in spite of new therapies. Nevertheless, the response to treatment was variable, mainly due to the heterogeneity of the disease. Long periods with diet alone have been observed in few cases. On the contrary, other patients were resistant to corticosteroids or immunosuppressive therapy. Cyclosporine induced remission in the majority of cases, and later, the treatment with tacrolimus was reported to be successfully in patients previously resistant to other immunotherapies.
IPEX syndromePowell et al.15 reported the first case of IPEX syndrome. This rare disease is characterized by immune dysfunction, polyendocrinopathy, enteropathy and X-linked inheritance, which seems to be equivalent to the abnormalities expressed by the scurfy mice, also an X-linked abnormality which was accidentally produced in 1949.16,17 Before the accurate characterization, multiple terms were assigned to IPEX syndrome (Table 2).
Ancient terms assigned to IPEX syndrome (Marabelle et al. 2008)
| 1. X-linked Autoimmunity-Allergic Dysregulation Syndrome (XLAAD) |
| 2. Insulin-dependent Diabetes Mellitus Diarrhoea Syndrome (DMSD) |
| 3. X-linked Autoimmunity-Immunodeficiency Syndrome |
| 4. X-linked Diarrhoea, Polyendocrinopathy, Fatal Infection Syndrome |
| 5. Autoimmune Enteropathy with Haemolytic Anaemia and Polyendocrinopathy |
| 6. X-linked Polyendocrinopathy, Immune Dysfunction and Diarrhea (XPID) |
| 7. Diabetes Mellitus, Congenital Insulin-dependent with Fatal Secretory Diarrhoea |
| 8. Immunodeficiency, Polyendocrinopathy and Enteropathy, X-linked |
Symptoms of the IPEX syndrome are different from one patient to another; they can even vary within the same family, especially in severity of symptoms. The most common findings are diabetes mellitus, protracted diarrhoea, failure to thrive, eczema and haemolytic anaemia.17 Other less common features are thrombocytopenia with platelet-antibodies, autoimmune hypothyroidism and lymphadenopathy with splenomegaly.
- 1.
Gastrointestinal disease. It is characterized by a severe watery diarrhoea that could be also mucoid or bloody.18 Generally a worsening occurs when breast-feeding is switched to regular formula, and many patients develop food allergies, frequently associated to atopic eczema.19 The response to dietary manipulation is variable, and symptoms may persist, requiring total parenteral nutrition to reverse the failure to thrive.
- 2.
Autoimmune endocrinopathies. The onset is very early and the involvement of pancreas and thyroid is the most common feature of IPEX syndrome, whereas the involvement of parathyroids, or adrenals is rare. Insulin-dependent type 1 diabetes begins frequently during the first year of life, although in some cases, glucose intolerance is already present at birth. Thyroiditis is common and can lead to hypothyroidism or hyperthyroidism.
- 3.
Dermatitis. A mild eczema is the most common finding in the skin and it may onset as an erythematous rash covering all the body.18 Other immune-mediated dermatological diseases described are pemphigoid nodularis,20 psoriasiform dermatitis or alopecia. Patients with lesions resembling onychomycosis were also published.21
- 4.
Other findings. In addition to the basic triad, other autoimmune alterations were reported in patients with IPEX syndrome. The most common are autoimmune haematological disorders, such as Coombs-positive haemolytic anaemia, thrombocytopenia or neutropenia, which altogether are present in more than 50% of the patients. Renal disease affects about 30% of the cases, mainly as an interstitial nephritis, although membranous glomerulonephritis was also reported.22 The severity of the clinical picture is variable; ranging from mild proteinuria and haematuria to rapidly progressive glomerulonephritis, only reverted by stem cell transplantation.22 Autoimmune hepatitis is also common. Some patients have neurologic abnormalities with developmental delay or seizures.
A high susceptibility to infections and reactions to vaccines was denoted in many patients without an apparent immunodeficiency. Some authors related infections to alterations of the skin and the gut barrier, or to immunosuppressive therapy. The neutropenia may be another influent factor.
Until now, only cases with typical IPEX syndrome have been easily identified, however cases with mild and uncompleted clinical pictures are increasingly reported. Two unrelated cases with early onset enteropathy and long-term survival, without type 1 diabetes, were recently published. They showed a novel mutation of FOXP3 gene.23
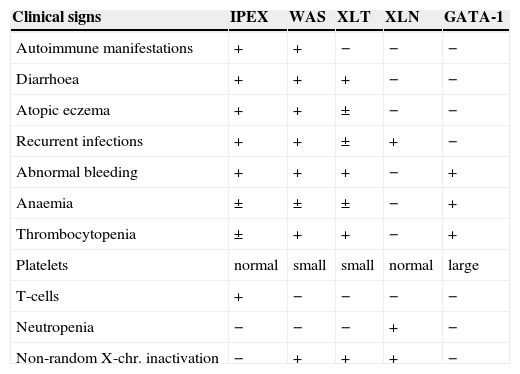
The definitive diagnosis is based on genetic studies and the identification of the mutation, but an easier screening test by immunocytochemical staining of FOXP3 molecule in bowel biopsies has also been proposed.24 The Wiskott-Aldrich syndrome and other X-linked diseases with immune abnormalities have some similarities (Table 3). Although previously a common genetic origin was proposed for all these entities, definitively a separate identity has been proven for every syndrome.17
Differential characteristics of X-linked syndromes with immune abnormalities (Bennett et al. 2001)
| Clinical signs | IPEX | WAS | XLT | XLN | GATA-1 |
| Autoimmune manifestations | + | + | − | − | − |
| Diarrhoea | + | + | + | − | − |
| Atopic eczema | + | + | ± | − | − |
| Recurrent infections | + | + | ± | + | − |
| Abnormal bleeding | + | + | + | − | + |
| Anaemia | ± | ± | ± | − | + |
| Thrombocytopenia | ± | + | + | − | + |
| Platelets | normal | small | small | normal | large |
| T-cells | + | − | − | − | − |
| Neutropenia | − | − | − | + | − |
| Non-random X-chr. inactivation | − | + | + | + | − |
IPEX: Immunodysregulation, polyendocrinopathy, enteropathy, X-linked.
WAS: Wiskott-Aldrich syndrome.
XLT: X-linked thrombocytopenia.
XLN: X-linked neutropenia.
GATA: is a T-cell transcription factor.
Our knowledge about immune findings is fragmented and incomplete, even contradictory, because infants are frequently studied during serious illness, malnutrition, infections or immunosuppressive therapy No patognomonic abnormalities and consistent changes in number of cells or immune function have been described.25 Nevertheless, IgE serum levels are increased in most patients with IPEX syndrome, and eosinophilia and high IgA are also common. Serum anti-islet, thyroid cell or enterocyte antibodies are present in some patients. Most patients have normal peripheral blood number of CD3+ CD4+ and CD8+ T-cells, with normal proliferative response to mitogens. Some patients showed a low number of NK and B cells, in this case with an associated hypogammaglobulinaemia and recurrent bacterial infections.26 The constant finding is the depletion of naive T-cell (CD4+ and CD8+) with increase number of memory T cells.26,27 A high production of different cytokines, lymphocyte strong response to mitogens and increased expression of MHC class II suggest a global activation of the immune response.17
A severe villous atrophy accompanied by lymphocytic infiltrates of lamina propria, mucosa and submucosa are characteristic findings of IPEX syndrome. Lymphocytic inflammation of pancreas with complete destruction of islet cells is present in cases with diabetes.17 The typical lymphocytic infiltration may also involve other organs, such as thyroid, skin, liver or brain.
The IPEX syndrome shares features with other X-linked syndromes, such as Wiskott-Aldrich syndrome, X-linked thrombocytopenia or X-linked congenital neutropenia. Genetic studies have clearly defined the IPEX syndrome as a different entity.
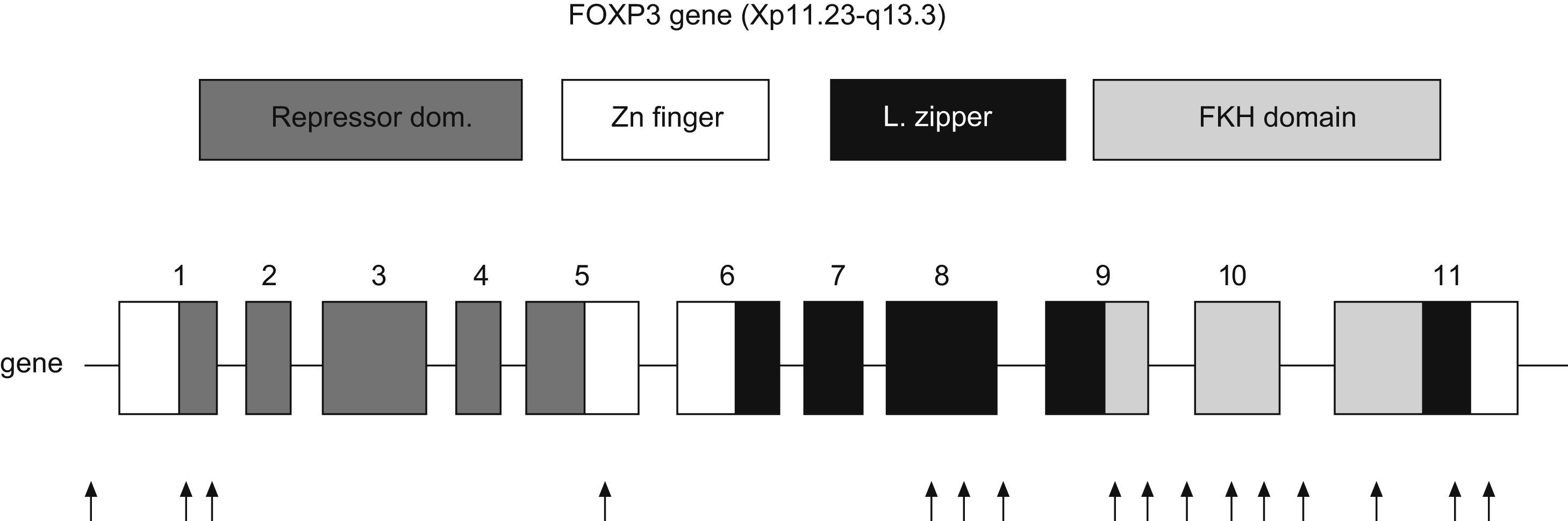
FOXP3The FOXP3 gene is located in the short arm of chromosome X (Xq11.23-Xq13.3). It has 11 exons which encode a protein of 431 amino acids. It is expressed predominantly, but not exclusively, in CD4+CD25+ T-cells. Transcription factors are proteins which bind regulatory regions of DNA and augment or suppress the transcription of particular genes. FOXP3 is a member of subfamily of forkhead box (FOX) transcription factors, which play a predominant role in embryonic pattern, development and metabolism.28
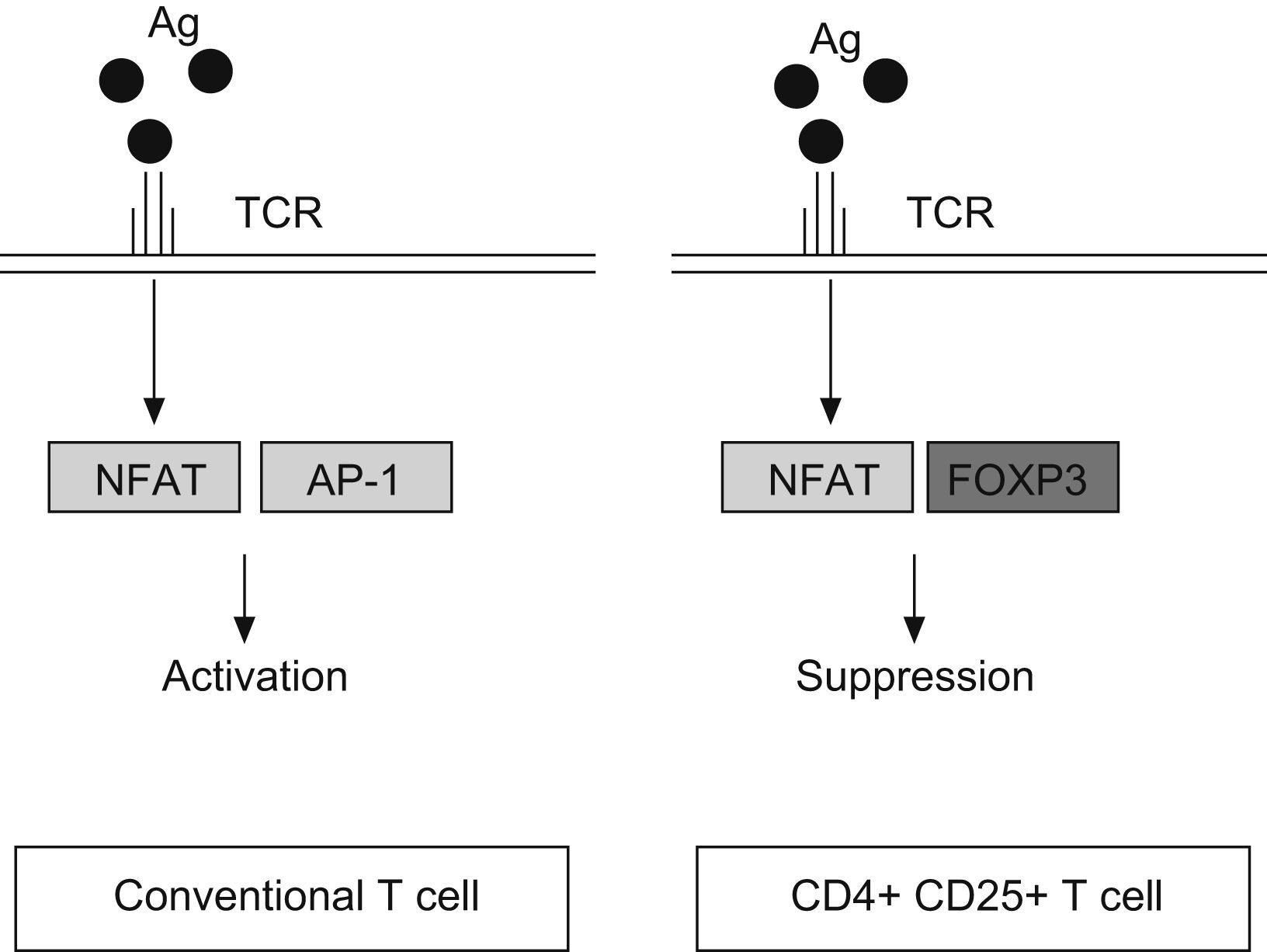
FOXP3 represses the promoter of several cytokine, such as IL-2 or GM-CSF. It interacts with the nuclear factor of activated T cells (NFAT) and perhaps with acute myeloid leukaemia 1/runt related transcription factor (AML1/Runx1) and nuclear factor-kB, forming complexes that repress different cytokines promoters29–31 (Fig. 2). The binding of NFAT with FOXP3 occurs specifically in CD4+CD25+ Treg cells, whereas in conventional T cells the NFAT exerts the repression function binding through another transcription factor, the activator protein-1 (AP-1).25
.")
Representation of human FOXP3 gene. Repressor domain binds NFAT molecule, Zn finger with leucine zipper domain promotes the oligomerization of FOXP3 molecule and its function; Forkhead domain binds DNA sequence. Arrows indicate the location of mutations in patients with IPEX syndrome (Modified from van der Vliet et al. 2007).
Three domains are key for the function of FOXP3: a C-terminal region which contains the forkhead domain that directly binds DNA regions; a central domain with a zinc finger and a leucine zipper that promotes the oligomerization of the FOXP3 molecule32; and a repressor domain located in the N-terminal region which binds the NAFT (nuclear factor of activated T cells) molecule33 (Fig. 3).
 up-regulates the expression of nuclear factor of activated T cell (NFAT) both, in conventional T cells and CD4+ CD25+ T cells. However, NFAT binds the activator protein 1 (AP1) in conventional T cells, whereas in Treg (CD4+CD25+) reacts with FOXP3 factor. It produces the transcription of a different set of genes, resulting in a final activation reaction in T conventional cells and suppression in Treg cells.")
Antigen reaction with T cell receptor (TCR) up-regulates the expression of nuclear factor of activated T cell (NFAT) both, in conventional T cells and CD4+ CD25+ T cells. However, NFAT binds the activator protein 1 (AP1) in conventional T cells, whereas in Treg (CD4+CD25+) reacts with FOXP3 factor. It produces the transcription of a different set of genes, resulting in a final activation reaction in T conventional cells and suppression in Treg cells.
Transgenic mice with increased expression of FOXP3 show decreased number of CD4+ T cells, a low cellularity in lymphoid tissues and a poor response to antigenic stimulation.28 Some cytokines (TGF-beta and IL-2), and the stimulation of TCR, have been identified as factors that induce the expression of FOXP3, although their regulation is poorly known yet.
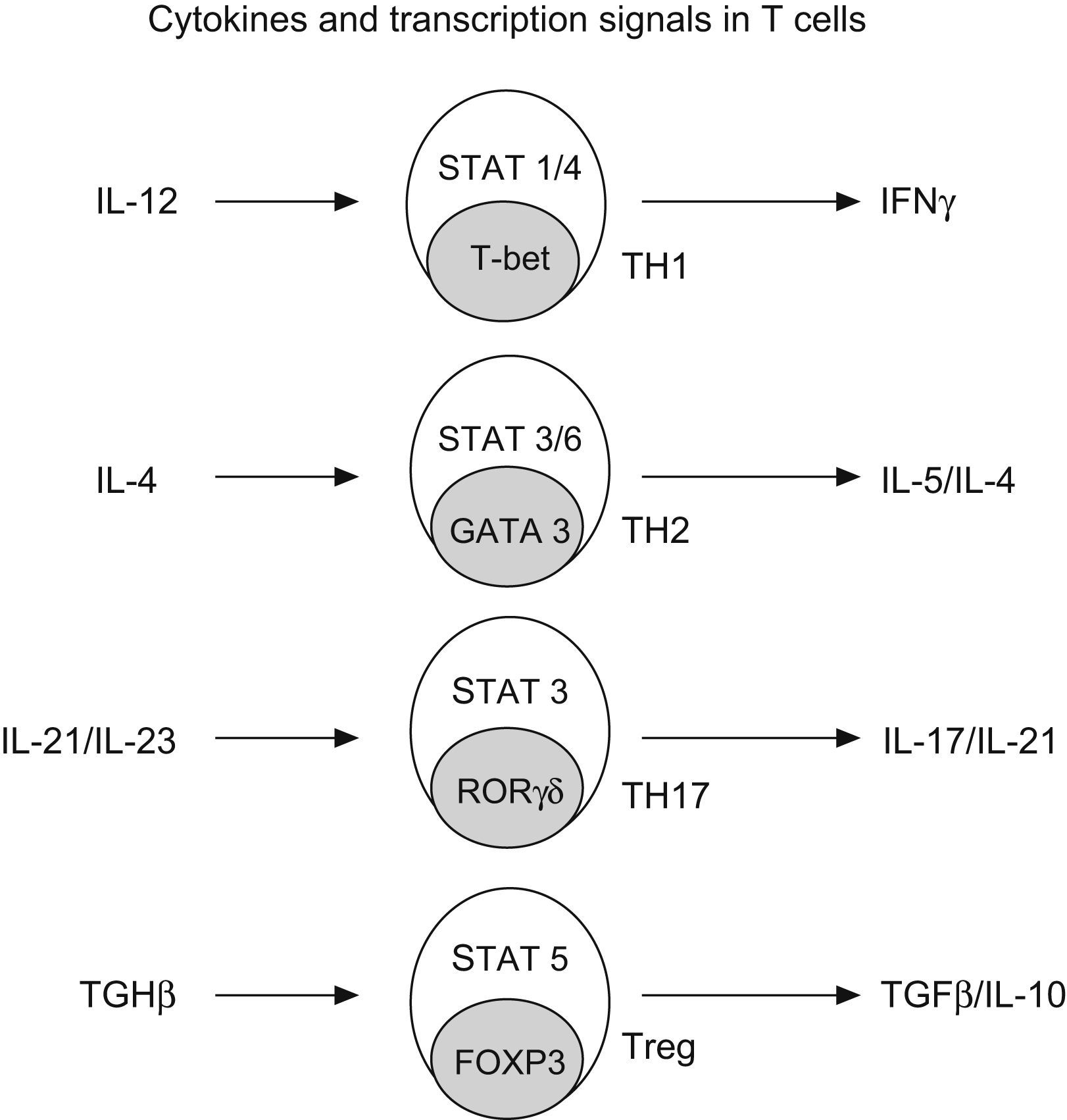
Regulatory T cells (Treg)Regulatory T cells constitute a small subset (5–10%) of CD4+ T helper cells that develop in the thymus. They are characterized by the expression of IL-2 receptor alpha chain (CD25), also expressing glucocorticoid-induced TNF receptor (GITR) and cytotoxic T lymphocyte-associated antigen 4 (CTL-4)25 (Fig. 4).
 and the nuclear factor FOXP3. The early production of IL-4 favours the development of Th2 by STAT 3/6 and GATA3; IL-12 promotes TH-1 by STAT1/4 and t-bet; and IL-21 and IL-23 promotes TH-17 by STAT 3 and RORgt (Modified from Bacchetta et al. 2007).")
Generation of CD4+ subtype cells. Transforming growth factor B-1 promotes the development of Treg cells by signal transducer and activator of transcription 5 (STAT5) and the nuclear factor FOXP3. The early production of IL-4 favours the development of Th2 by STAT 3/6 and GATA3; IL-12 promotes TH-1 by STAT1/4 and t-bet; and IL-21 and IL-23 promotes TH-17 by STAT 3 and RORgt (Modified from Bacchetta et al. 2007).
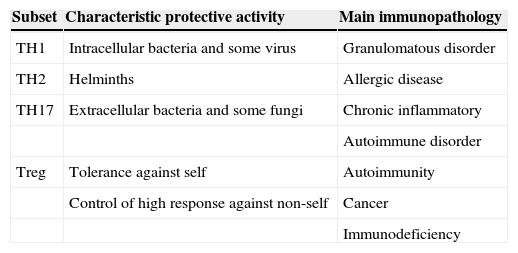
Treg cells are of critical importance for the establishment of the self-tolerance and immune homeostasis.34 It has been shown that these cells suppress the autoimmune, the antimicrobial and the antitumoral responses25 (Table 4). More recently, a novel anti-atherogenic role was claimed for Treg.35 It is noteworthy that antigen simulation is required for the initiation of Treg suppression, but suppressive effects can later occur to bystander cells and systems. They play an important role in controlling exaggerated Th2 response and they are involved in cow's milk allergy as well as in the allergy to inhalants.36,37 The capacity of Treg to induced oral antigen tolerance has been proven in mice38–40 and is also supposed in man. A defect Treg function was associated to autoimmune, allergic and immunodeficient diseases.41,42 In contrast, an increased Treg function has been associated to malignancies.43–46
Simplified view of protective role and immunopathology involvement of different CD4+ T-cell subset (Romagnani 2006)
| Subset | Characteristic protective activity | Main immunopathology |
| TH1 | Intracellular bacteria and some virus | Granulomatous disorder |
| TH2 | Helminths | Allergic disease |
| TH17 | Extracellular bacteria and some fungi | Chronic inflammatory |
| Autoimmune disorder | ||
| Treg | Tolerance against self | Autoimmunity |
| Control of high response against non-self | Cancer | |
| Immunodeficiency |
FOXP3 plays a crucial role in the generation of Treg, and when it is absent, these cells do not develop in mice nor in man. Transgenic mice (scurfy) which do not express FOXP3 lack Treg. To date, about 20 mutations of FOXP3 have been identified. The majority are missense mutations that affect the integrity of the forkhead domain, but deletions and substitutions have also been identified in the leucine zipper and in the repressor domain. Nevertheless, the genetic basis o the disease seems to be more heterogeneous than initially presumed.
The forkhead mutations interfere with nuclear import and DNA binding. Mutations located in N terminal region affect the regulation of FOXP3 on the NFAT protein. Other mutations seem to increase the length of the FOXP3 protein, changing the three-dimensional structure and the function.25 In some cases, no mutations in gene FOXP3 are identified and it is supposed that they involve some unknown regulator gene which may be located even outside the X chromosome, because females are affected in some families.44,47,48 Moreover, a case with gene FOXP3 mutation, normal FOXP3 protein expression and Treg deficient function was published, proving that the complex mechanism of this disorder is yet unclear.25,49 Some reports support a genotype-phenotype correlation.50
Treatment of IPEX syndromeSymptomatic treatment includes total parenteral nutrition and insulin injections that are commonly needed, as well as red blood or platelet transfusion. In the early years, the therapy of IPEX syndrome was based on immunosuppressive drugs. Steroids, methotrexate, cyclosporin, FK506 and more recently infliximab and rituximab have been used with unequal results, frequently disappointing, because the response is short and late complications may appear. Sirolimus is the latest drug assayed and seems to be less nephrotoxic and better tolerated, improves diarrhoea and villous histology, and decreases the dosage of corticoids. In addition, it allows the expansion of Treg while the growth of effector T cells is inhibited.51,52
In spite of new drugs, bone marrow transplantation is the only definitive cure for patients with IPEX syndrome.53 It has been performed in some cases with complete clinical remission, in spite of only a partial engraftment with a chimerical status.54 After transplantation, clinical recovery was associated with the emergence of regulatory T cells, the majority expressing markers of memory phenotype.53 It was remarkable that the conditioning regimen was itself effective in controlling symptoms in some patients and it was recently noticed that a reduced-intensity conditioning regimen prior to the engraftment resulted in more FOXP3T regulatory cells than more aggressive conditioning.55 Correction symptoms were also successfully reached with cord blood donor.56 Unfortunately, the excellent results of bone marrow transplantation are frequently transient.17
The recovery of diabetes mellitus was obtained in some cases, but in most instances the islet damage is already permanent at the time of bone marrow transplantation.28 It is recommended to perform the transplantation in the early phase of the disease in order to avoid irreversible damage and complications.
Complications. Malnutrition is the main complication in infants, and infections in later ages, being the most frequent cause of death. Lymphoproliferative haemophagocytic syndrome is another complication which was also published.47,54 The immunosuppressive drugs and their dose are one of the most important risk factors of complications, so the type of therapy must be carefully considered. A patient with IPEX syndrome managed with rapamycin who subsequently developed a lymphoma induced by Epstein Barr virus was recently reported.57
IPEX-like syndromesDue to the complexity of Treg function it is not surprising to find patients with similar symptoms and different genetic and immune findings.
CD25 deficiency. From the discovery of IL-2R alpha (CD25) defects in 1997,58 at least two families were described with a genetic mutation and a lack expression.59 The main characteristic data was a massive enlargement of peripheral lymphoid organs, with hepatosplenomegaly, enlarged tonsils and lymphadenopathy. All cases show an inflammatory bowel disease with villous atrophy. One case had clear traits of immunodeficiency, with low T cell number and decreased response to mitogens whereas the second patients showed an IPEX typical pattern with several autoantibodies. It was remarkable that the first patient did not develop diabetes or endocrinopathies, and the diabetes showed a late onset, 6 year of age, in the case with IPEX pattern.60,61
In summary, patients who years ago were diagnosed as AIE may be now considered to have IPEX syndrome. The majority of cases could be confirmed by studying FOXP3 gene mutations. Nevertheless, there are also atypical cases which have appeared with a delayed onset, or affecting female patients. They may be variants of FOXP3 mutation or be related to other molecules also involved in the generation or the function of Treg cells, such as CD25 abnormalities.
Conflict of interestThe authors declare that they have no conflict of interest.
