


Susceptibility to infections caused by Mycobacterium tuberculosis and other Mycobacterium species has been reported in patients with acquired immunodeficiencies (e.g., acquired immunodeficiency syndrome, or AIDS) or primary immunodeficiency diseases (PIDs), including defects of the IL-12/IFN-γ axis, severe combined immunodeficiency (SCID), CD40L deficiency, and chronic granulomatous disease (CGD). The latter is caused by genetic defects that affect the NADPH oxidase in phagocytes, which results in the failure of neutrophils and monocytes/macrophages to generate reactive oxygen species and, hence, an inability to kill pathogens, including fungi and intracellular bacteria.1
The phagocyte NADPH oxidase complex consists of two membrane-bound proteins (gp91phox and p22phox), which together form cytochrome b558, and three cytosolic proteins (p47phox, p67phox, and p40phox). Upon cell activation, these proteins and the small GTPase Rac assemble at the cell membrane and at phagolysosomes and trigger the oxidative burst, which is essential for microbial killing. Mutations in genes encoding components of NADPH oxidase have been described in CGD.2 Among the genes encoding the NADPH oxidase subunits, mutations in CYBB, which encodes gp91phox and is located on the X chromosome, account for approximately 65% of CGD cases, whereas mutations in autosomal genes, such as CYBA (encoding p22phox), NCF1 (encoding p47phox), NCF2 (encoding p67phox), and NCF4 (encoding p40phox), cause autosomal recessive (AR)-CGD. The most common form of AR-CGD is caused by mutations in NCF1 (25% of CGD cases), followed by rare mutations in NCF2, CYBA, and NCF4.
Patients with CGD suffer from recurrent, life-threatening bacterial and fungal infections that affect the skin, the lungs, the liver, the spleen, lymph nodes, and bones. In North America and Europe, the most commonly observed infections in CGD are caused by Staphylococcus aureus, Burkholderia cepacia, Salmonella sp., Serratia marcescens, Nocardia spp., and Aspergillus spp. In addition to these pathogens, CGD patients may develop TB and clinical adverse reactions following Bacille Calmette–Guerin (BCG) vaccination (BCGites or BCGosis), particularly patients in regions with a high incidence of TB, such as South America and Asia.3
Mycobacterial infections have been reported in patients with defects in gp91phox, p22phox, and p67phox.4 However, to the best of our knowledge, we describe the clinical and molecular features of the first patient with severe M. tuberculosis infection (TB) and a defect in p47phox (p.Val25X51). In addition, a defective immune response to M. tuberculosis from the patient's monocyte-derived macrophages is characterized.
The female patient was born in 1997 to non-consanguineous Brazilian parents after an uncomplicated pregnancy. Her mother died of an unknown cause, and her father is alive and healthy. The patient was vaccinated with BCG without clinical complications. She developed pneumonia at eight months of age and remained without clinical manifestations until 6 years of age. At this age, she developed pneumonia with pleural empyema, granulomatous pleuritis, osteomyelitis of the rib, and septicaemia with a poor response to different antibiotic regimens. A lung biopsy showed pulmonary mycosis due to Aspergillus sp. and Zygomycetes, the species of which could not be identified. These fungal infections were successfully treated with amphotericin B. One year later, the patient presented with other severe infections, including meningitis, endocarditis, and pulmonary mycosis, with a positive Candida albicans culture. The laboratory results revealed normal leucocyte counts and normal serum IgA, IgM, and IgG levels. At 12 years of age, the patient developed persistent fever, dry cough, and weight loss. A computed tomography (CT) scan and chest radiograph suggested severe TB, and M. tuberculosis was identified in the lung biopsy (Fig. 1A) and biopsy specimen culture. To control the TB clinical symptoms, the patient needed a longer course of treatment (3 years) than usual (Ministry of Health of Brazil Guidelines – http://bvsms.saude.gov.br/bvs/publicacoes/tratamento_diretamente_observado_tuberculose.pdf) with rifampicin, isoniazid, and pyrazinamide. Currently, the patient is receiving prophylactic cotrimoxazole and itraconazole.
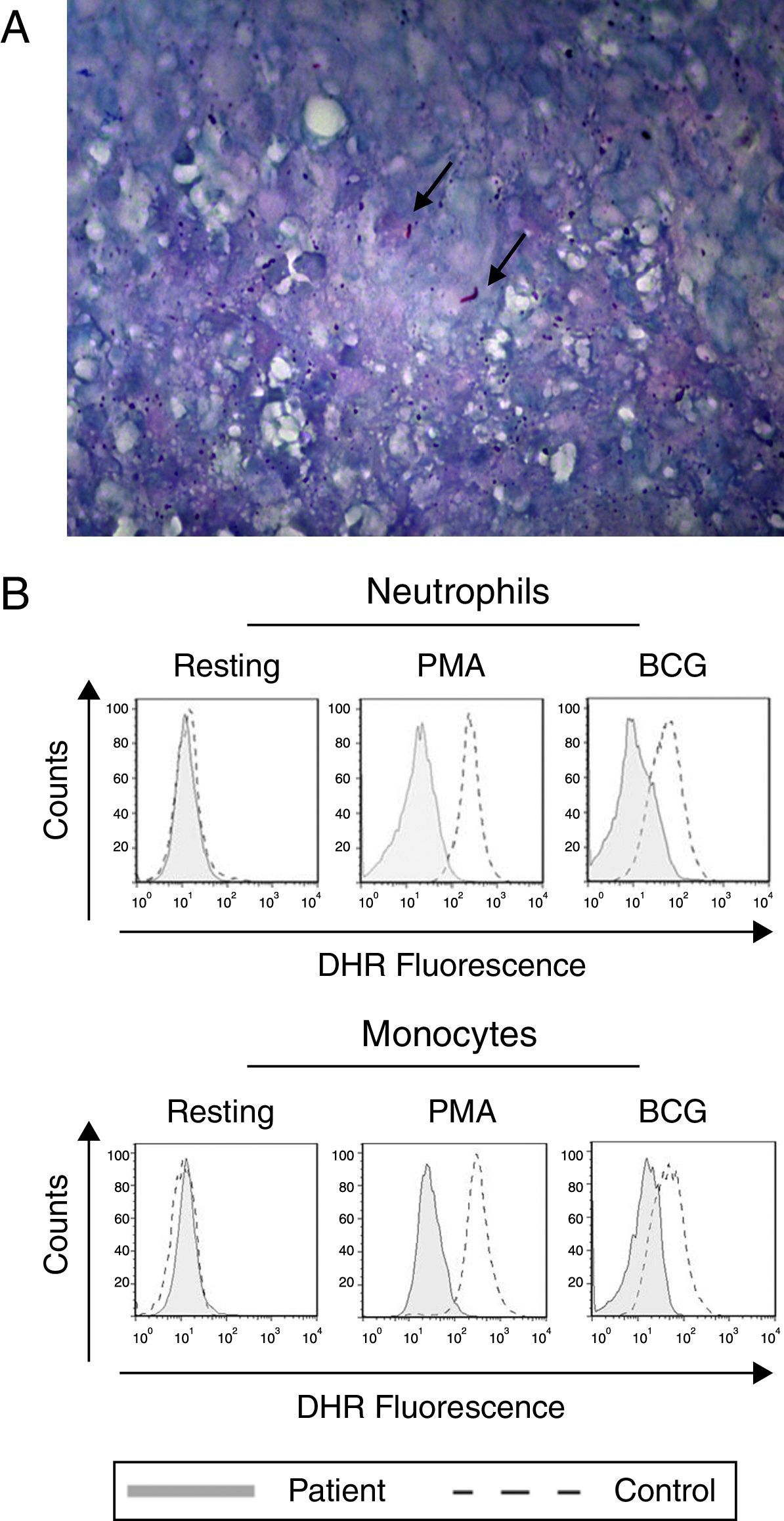
 The lung biopsy specimen shows the presence of M. tuberculosis. The Ziehl–Neelsen-stained photomicrograph shows the presence of Mycobacterium tuberculosis (arrow). (B) Neutrophils and monocytes from our index case show a reduced oxidative burst after stimulation with PMA or BCG as shown in the DHR assay.")
Tuberculosis and defective oxidative burst. (A) The lung biopsy specimen shows the presence of M. tuberculosis. The Ziehl–Neelsen-stained photomicrograph shows the presence of Mycobacterium tuberculosis (arrow). (B) Neutrophils and monocytes from our index case show a reduced oxidative burst after stimulation with PMA or BCG as shown in the DHR assay.
Considering the patient's medical history, she was investigated for a defect in the IL-12/IFN-γ axis and for CGD. The laboratory investigation was approved by the Ethics Committee at the Institute of Biomedical Sciences, University of São Paulo, according to the Helsinki Convention and the Ministry of Health of Brazil. Informed consent was signed before the laboratory examinations were performed. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood by Ficoll–Hypaque sedimentation and analysed for the production of IL-12 and IFN-γ, as previously described.5 The oxidative burst was analysed using the dihydrorhodamine 123 (DHR, Sigma–Aldrich) assay, and the expression of phagocyte NADPH oxidase components was detected by flow cytometry. The patient's genetic defect was determined by genetic sequencing of the NCF1 gene, including its exon–intron boundaries. Human monocyte-derived macrophages (MDMs) were generated, and bacterial uptake (phagocytosis) data were obtained from the colony-forming unit (CFU) counts performed on day 0. The bacterial proliferation index was determined based on the ratio of the CFU numbers on days 3 and 6 compared to the CFU number on day 0. Detailed material and methods are available upon request.
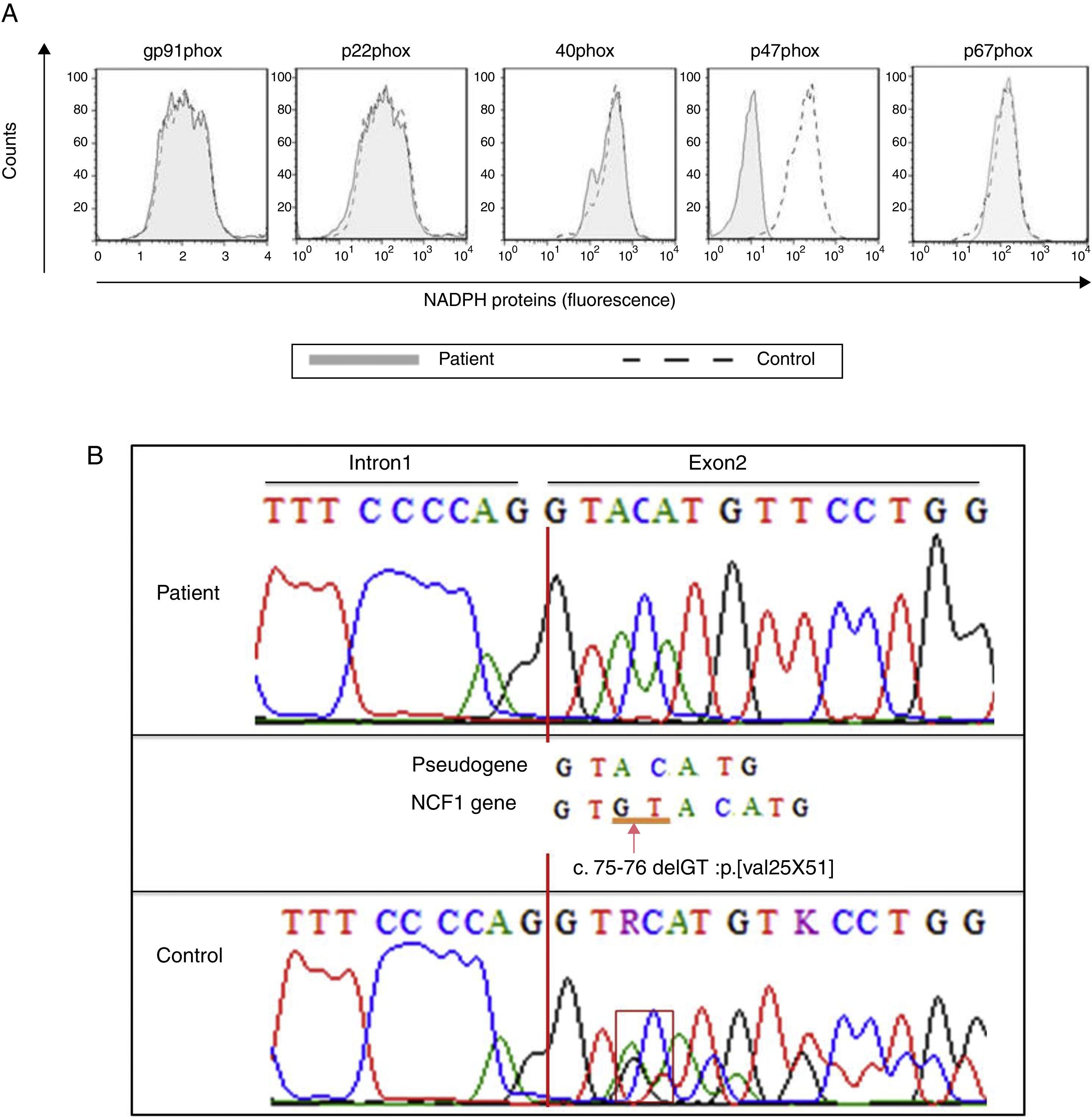
The production of IL-12 and IFN-γ in response to all stimuli was normal compared with healthy controls (data not shown). In contrast, the evaluation of the oxidative burst response using the DHR assay revealed that neutrophils and monocytes from the patient presented impaired oxidative burst in response to phorbol 12-myristate 13-acetate (PMA) and BCG (Fig. 1B) compared with phagocytes from healthy controls. Taken together, these data suggest CGD. Compared with monocytes that were obtained from healthy subjects, monocytes from the patient displayed normal expression of all NADPH subunits, except p47phox, which was reduced (Fig. 2A). Sequencing of the NCF1 gene identified the mutation g.3307_3308delGT, which is a well-known homozygous frameshift GT deletion (c.75–76delGT; g.3307_3308delGT; p.Val25X51) located at the beginning of exon 2 (Fig. 2B), which underlies the CGD phenotype.
 Histograms show defective p47phox expression in the patient's monocytes. (B) Sequencing shows the homozygous deletion (g.3307_3308delGT) identified in the gDNA of our index case.")
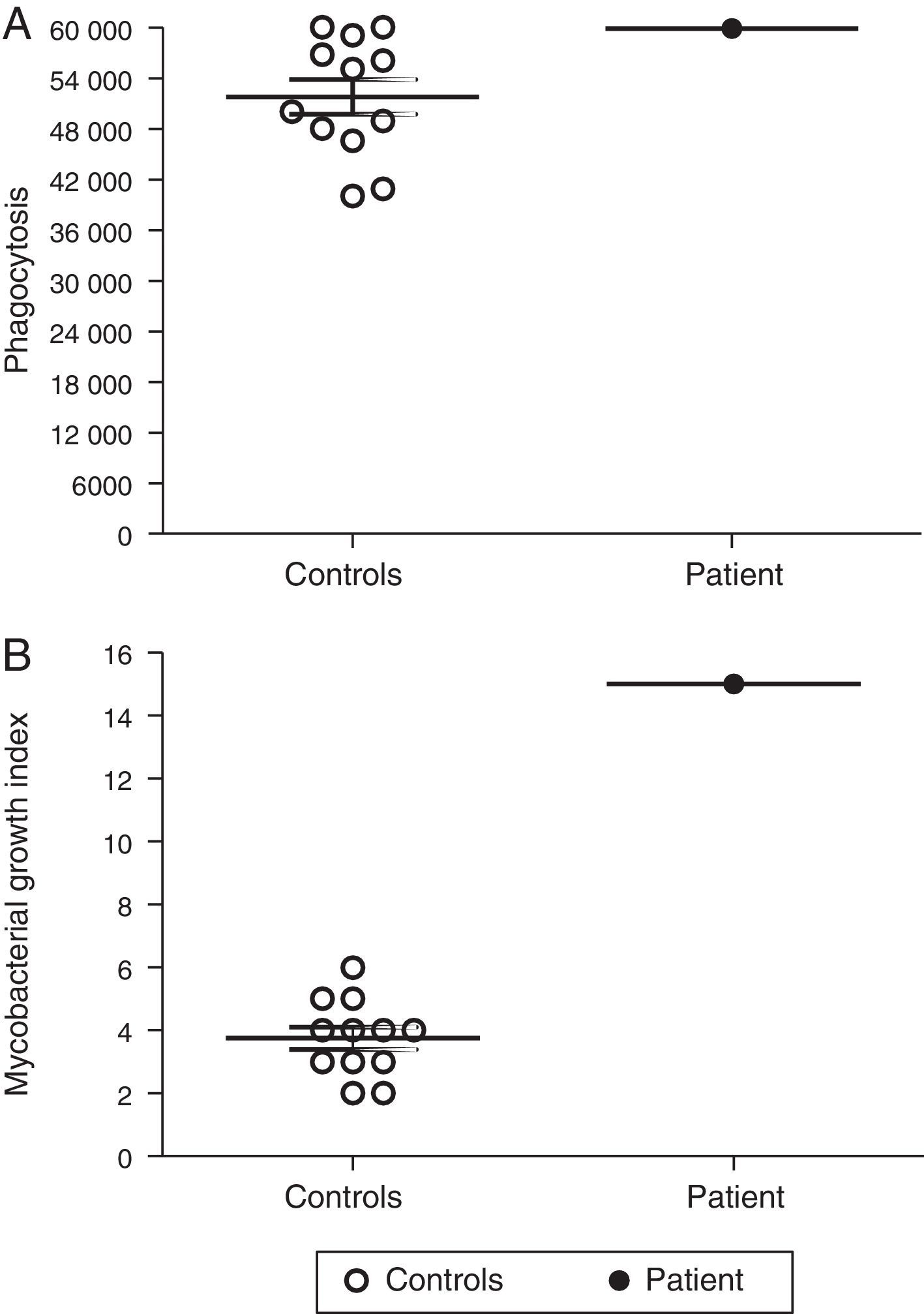
Macrophages are essential for immune protection against TB by controlling M. tuberculosis proliferation. Therefore, we analysed the capacity of MDMs from our p47phox-deficient patient to phagocytose and to control intracellular M. tuberculosis proliferation. The ability of MDMs from the p47phox-deficient patient to phagocytose M. tuberculosis was similar to that of healthy controls (Fig. 3A). However, the patient's MDMs failed to kill M. tuberculosis (Fig. 3B).
, and the bacterial proliferation index was determined. The patient's MDMs exhibited normal phagocytosis but failed to control the proliferation of the bacilli. The results are representative of two independent experiments that were performed in triplicate.")
Defective control of M. tuberculosis proliferation by MDMs from the p47phox-deficient patient. Macrophages were cultured with Mycobacterium tuberculosis (H37Rv strain), and the bacterial proliferation index was determined. The patient's MDMs exhibited normal phagocytosis but failed to control the proliferation of the bacilli. The results are representative of two independent experiments that were performed in triplicate.
The spectrum of infections observed in CGD patients is broader than those in patients with Mendelian susceptibility to mycobacterial diseases (MSMD), which are associated with defects of the IL-12/IFN-γ axis. In addition to mycobacterial infections, patients with CGD are highly susceptible to multiple life-threatening bacterial and fungal infections.4 Accordingly, our patient presented a typical CGD clinical phenotype and the common GT deletion in NCF1, which is the unique hot-spot mutation in all CGD forms.6
Brazil has one of the highest rates of TB infections when compared with other countries where BCG vaccination is a mandatory practice. However, BCG vaccination can cause clinical complications (BCGitis/BCGosis) in CGD patients, including those with p47phox deficiency. Nevertheless, despite receiving a BCG vaccine without clinical complications, patients with any CGD form can fail to develop a protective immune response against Mycobacterium species and may develop TB later in life, which was the case in our patient. However, to date, 72 patients with CGD have been described with M. tuberculosis infections, whereas 220 CGD patients developed BCG complications, which suggests that there is no correlation between the occurrence of BCG complications and M. tuberculosis infections.
Notably, approximately 20% of the published cases of mycobacterial infections in all CGD forms are caused by M. tuberculosis.4 However, M. tuberculosis infection has not been described in patients with mutations in the NCF1 gene most likely due to the limited number of well-established laboratories that perform molecular testing in developing countries where TB is still endemic.7
Immunity to mycobacteria has been ascribed to T-cell-mediated immunity. The T-cell immune response plays a crucial role by producing IFN-γ, which increases the oxidative burst of phagocytes by modulating NADPH activation.8 Macrophages have been described as the main effector cells that kill M. tuberculosis.9 Accordingly, we have demonstrated for the first time that MDMs from a p47phox-deficient patient exhibit a normal capacity to phagocytose M. tuberculosis but fail to control the intracellular growth of M. tuberculosis a defect that has been displayed by p47phox knock-out mice.4
In conclusion, our data emphasise the relevance of early molecular genetic diagnosis of CGD and suggest that patients with mutations in the NCF1 gene need to be monitored for M. tuberculosis infections. Additionally, this work highlights the important role of the phagocyte NADPH oxidase in the immune response of MDMs against M. tuberculosis.
Ethical disclosuresConfidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
The authors would like to thank Dunia Rodriguez and Luciana Cezar de Cerqueira Leite from the Butantan Institute Sao Paulo, Brazil, and Dr. Philip Suffys from the Fundação Oswaldo Cruz (FIOCRUZ, Brazil) for kindly providing BCG and M. tuberculosis specimens, respectively. This work was funded by the Fundaçãode Amparo a Pesquisa do Estado de São Paulo (Fapesp 10/51814-0, 13/50303-0, 2012/50515-4) and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 470527/2014-5). TAK was supported by The World Academy of Sciences (TWAS).
