




La clasificación de las inmunodeficiencias primarias (IDP), que comenzó a publicarse en 1971 bajo el auspicio de la OMS, se actualiza periódicamente y resume el estado del arte en el conocimiento de estas enfermedades. La última se ha publicado en 1999 (1) y como es habitual presenta algunas novedades. La primera de ellas, es que por primera vez es la Unión de Sociedades de Inmunología (IUIS) quien la patrocina. La segunda es la creación de un nuevo grupo de IDP, el de las inmunodeficiencias asociadas a trastornos proliferativos, que se añade a los siete grupos de la clasificación anterior (tabla I). Además, en grupos como el de las inmunodeficiencias combinadas o en el de las calificadas como bien definidas se aumenta el número de entidades. Este aumento se debe a la reubicación de IDP ya conocidas, pero en las que nuevos datos sobre su etiología aconsejan un cambio de grupo. En otros casos son desdoblamientos de síndromes ya descritos en los que se ha descubierto que pueden ser producidos por más de un defecto genético y en algún otro a la definición de nuevos cuadros clínicos. Se sigue conservando el grupo de «otras inmunodeficiencias primarias» que agrupa a cinco inmunodeficiencias combinadas o fundamentalmente celulares de etiología incierta, sin caracterización a nivel génico y por lo general de incidencia muy escasa. También se conserva el grupo de inmunodeficiencias asociadas o secundarias a otras enfermedades congénitas o hereditarias, grupo también variopinto y no muy bien definido, en el que siguen ubicándose dos IDP que podríamos denominar clásicas: el síndrome de hiper-IgE y la candidiasis mucocutánea crónica.

Actualmente el progreso en el conocimiento de estas enfermedades es tan rápido, gracias sobre todo al desarrollo de la biología molecular, que desde la publicación de esta clasificación hasta el final del año 2000 se han descrito como mínimo, tres nuevas IDP: la deficiencia de la cadena a del receptor de la IL-7 (2), la deficiencia en CD45 (3) y la deficiencia en CD8 (4). Esta última descrita por una de las participantes en esta mesa, la Dra. Español. Es probable además, que las expresiones defectuosas del p55 lck y de la cadena b común de IL-2 e IL-15, den lugar a formas graves de inmunodeficiencias combinadas (5, 6). Es seguro que en los próximos años asistiremos a la descripción de nuevos defectos genéticos, sobre todo dentro de las inmunodeficiencias combinadas graves (IDCG), así como en las agammaglobulinemias autosómicas recesivas (7) y en los defectos micobactericidas de los leucocitos (8).
Otro de los cambios que se está produciendo en la clasificación de las IDP, es la inclusión de síndromes o enfermedades carentes de un síndrome infeccioso de repetición. Clásicamente se ha considerado que las IDP son enfermedades cuyo síntoma dominante son las infecciones repetidas, con frecuencia de especial gravedad, o producidas por microorganismos oportunistas. Cierto es que la deficiencia del inhibidor del componente C1 del complemento sérico, no origina sintomatología infecciosa y que la clínica de las deficiencias de los primeros componentes del complemento producen con mayor frecuencia enfermedades autoinmunes que infecciones y que en algunos pacientes afectos de forma variable común de inmunodeficiencia, la autoinmunidad predomina como manifestación clínica. Pero la inclusión de todas estas enfermedades en las anteriores clasificaciones de la OMS, parecía estar justificada porque, tanto en el conjunto de deficiencias de complemento como en el de los pacientes con forma variable común de inmunodeficiencia, las infecciones son el síntoma dominante. Pero en esta última clasificación se han incluido por primera vez los síndromes autoinmunes proliferativos, cuya clínica consiste en: linfoadenopatías, hepatoesplenomegalia, linfoproliferación policlonal de linfocitos T y B con aumento de los linfocitos CD3+ carentes de los antígenos de diferenciación CD4 y CD8 (células doble negativas). Además es constante la aparición de fenómenos autoinmunes y muy frecuente el desarrollo de enfermedades autoinmunes, sobre todo las dirigidas contra los elementos formes sanguíneos. Este síndrome se produce por un defecto en la apoptosis, bien por defectos genéticos en la expresión o función del Fas, de su ligando, de la caspasa 8 o por causas todavía desconocidas (9-11).
Da la impresión de que en un futuro, la clasificación de las IDP va a incluir a aquellos defectos genéticos de componentes del sistema inmune que originan manifestaciones patológicas por alteración en la respuesta inmune, sean o no estas manifestaciones infecciosas.
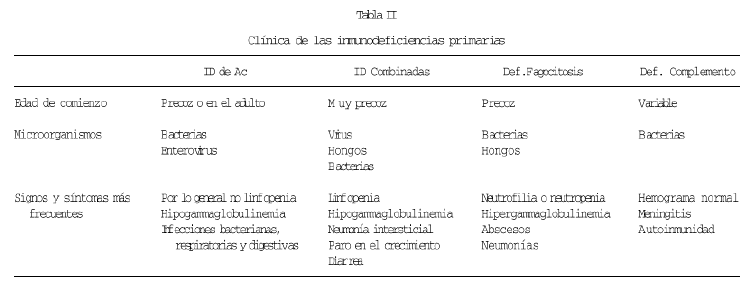
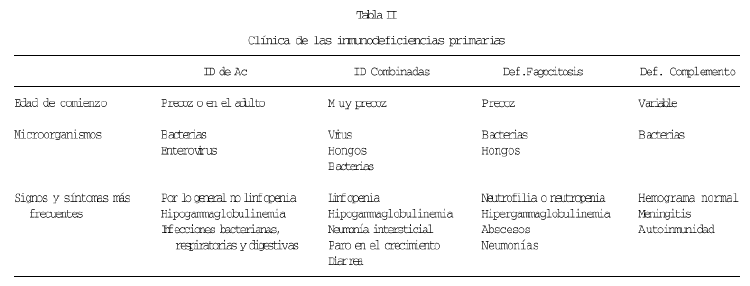
Por otra parte, el progreso en el conocimiento de la etiología y patogenia de estas enfermedades, está procurando un mejor diagnóstico y con ello un mayor conocimiento clínico de estos procesos. En cierto modo, se podría decir que se está ampliando el espectro sintomatológico. En la tabla II se resume la clínica de los cuatro tipos básicos de IDP, considerando como tipos básicos a los que afectan a cada una de las ramas clásicas de la inmunidad. Esta tabla es fundamentalmente correcta, pero alguno de estos aspectos están siendo revisados, o al menos, se están considerando las excepciones, que quizás sean más frecuentes de lo que pensamos. Vamos por ello a referirnos ahora a las llamadas formas variantes. Estas formas variantes con clínica o analítica atípica, tienen en algunos casos un correlato a nivel molecular, mientras que otras veces desconocemos su causa.
Un gen puede tener deleciones o mutaciones diversas que pueden, dependiendo de su localización, generar alteraciones cuantitativa y cualitativamente diferentes. Así puede no expresarse su producto, expresarse parcialmente o expresarse normalmente, lo que puede acompañarse de carencia de actividad o de actividad residual. Una actividad residual puede atenuar, o hacer diferente la expresividad clínica de una enfermedad. Esto puede explicar algunas de las formas variantes, pero en otros casos nos encontramos que una misma alteración génica, da manifestaciones en cierto modo distintas en personas afectas de una misma familia. En estos casos se sospecha una influencia significativa de factores ambientales o que la heterogeneidad de otros loci puedan modular la actividad del gen o de su producto.
Se considera que las IDP son mayoritariamente enfermedades que ven y diagnostican los pediatras y que son ellos los que deben conocer su clínica para poder sentar las bases diagnósticas. El síndrome variable común y las deficiencias de complemento, serían las excepciones a esta regla básica.
La agammaglobulinemia ligada al sexo (ALX) suele comenzar a dar síntomas hacia el año de vida, cuando los anticuerpos maternos transferidos por vía placentaria se agotan. Otra característica típica de esta enfermedad es la ausencia, prácticamente total, de todos los isotipos de inmunoglobulinas séricas, así como un porcentaje de linfocitos B circulantes inferior al 1 %. Esta es la regla a la que hace ya algún tiempo se conocen excepciones. Así, de los dos hermanos descritos por Bykowsky et al (12), el mayor tuvo una neumonía a los dos años de edad, pero posteriormente no presentó infecciones reseñables. Su hermano menor comenzó a los dos años a padecer sinusitis, otitis media y neumonías, lo que condujo a un retraso ponderoestatural grave. Se diagnostica de ALX típica y al hacer un estudio familiar se encuentra, secuenciando la Btk, que su hermano mayor, que tenía unas cifras de IgG e IgM normales, pero una deficiencia de IgA e IgG2, tiene también una ALX. En la familia descrita por Kornfeld (13), dos hermanos de 52 y 56 años de edad se diagnosticaron de ALX a partir de un estudio familiar por tener tres sobrinos diagnosticados de ALX. Existe un paciente descrito por Woods (14) que a los 6 y 17 años tuvo meningitis neumocócica. Tenía unas cifras de inmunoglobulinas normales, pero con una defectuosa producción de anticuerpos antipolisacáridos. Tras la secuenciación del gen de la Btk fue diagnosticado de ALX.
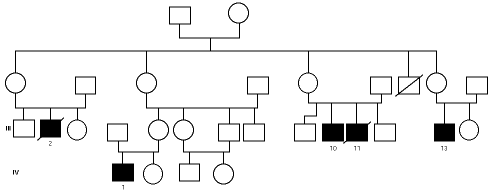
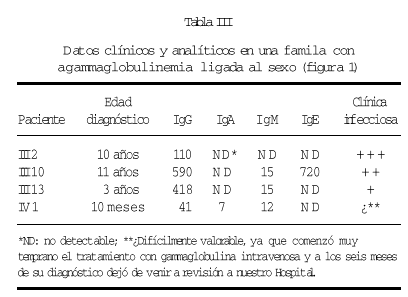
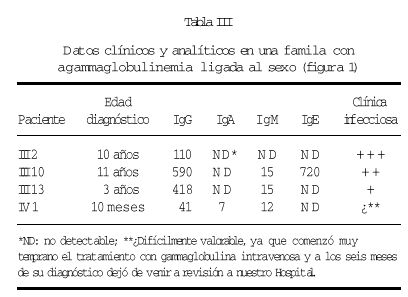
En una de nuestras familias (fig. 1), existen diferencias fenotípicas que se resumen en la tabla III. Todos los pacientes tienen idéntica mutación.
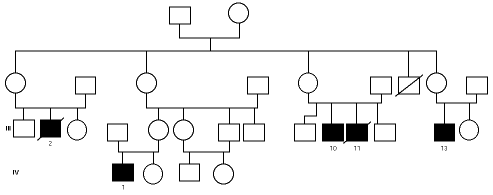
Figura 1.--Árbol genealógico de una familia con agammaglobulinemia ligada al sexo. Las características clínicas y analíticas se resumen en la tabla III.
Recientemente hemos visto a la hermana de una de nuestras pacientes diagnosticada a los 7 años de edad tras múltiples neumonías, probablemente afecta de una agammaglobulinemia autosómica recesiva, que presentó su primera manifestación clínica, una neumonía bilateral a los 16 años. Todos estos casos subrayan la necesidad de hacer estudios inmunológicos en los familiares de estos pacientes, tengan la edad que tengan y aunque no hayan padecido ninguna infección mayor.
Además no debemos olvidar que algunos pacientes diagnosticados de síndrome variable común con nulos o escasos linfocitos B circulantes, pueden ser en realidad ALX, o que otros con diagnóstico de ALX o forma variable común, pueden ser síndromes linfoproliferativos ligados al cromosoma X, lo que evidentemente puede variar su pronóstico (15).
Clásicamente se considera que la Inmunodeficiencia combinada grave (IDCG), suele tener un comienzo sintomatológico muy temprano, casi siempre antes de los 6 meses de vida y que el signo diagnóstico más útil es una linfopenia persistente. Pero en algunos casos no es así, lo que suele dificultar el diagnóstico.
La deficiencia en la enzima adenosinodesaminasa (ADA) da lugar a una forma de IDCG extraordinariamente linfopénica y de extrema gravedad. En los años 80 se describieron casos cuyo comienzo clínico no se producía hasta los 2-3 años de edad (16). En la década siguiente se publicaron dos hermanas cuya sintomatología no empezó hasta pasados los 20 años y que se diagnosticaron pasados los 30 (17). En 1997 se publicó una paciente de 39 años considerada sana hasta los 30 (18). Debido a estas características clínicas atípicas, los pacientes adultos con deficiencia de ADA, pueden ser diagnosticados de síndrome común variable o de linfopenia CD4 idiopática, lo que conlleva errores terapéuticos.
Otra forma de IDCG es la producida por mutaciones en el gen de la cadena g común del receptor de la IL-2. Estas pacientes, además de linfopenia presentan un fenotipo celular característico con ausencia de células T y NK, pero con células B circulantes.
Rodríguez Pérez y cols. (19), han estudiado seis pacientes con esta enfermedad, cinco de ellos tenían un fenotipo T + B + NK + , en dos de ellos debido a injerto de células maternas, pero en otros tres este fenotipo aparecía en ausencia de injerto. En todos los casos la gc se expresaba en la superficie celular. El sexto paciente que expresaba la gc con intensidad reducida, mostraba un fenotipo T-B + NK + . En uno de nuestros pacientes con deficiencia de gc, los linfocitos B no la expresaban, pero sí los monocitos y neutrófilos.
También se ha descrito a un paciente con una deficiencia de JAK3, deficiencia que da lugar a un fenotipo y clínica idénticas al de la deficiencia de gc, cuya sintomatología no dio comienzo hasta los 20 meses de edad y cuya cifra de linfocitos circulantes era normal, siendo las cifras de inmunoglobulinas séricas incluso elevadas para la edad del paciente. Además tenía tasas normales de isohemaglutininas y de anticuerpos antitetánicos (20).
Las deficiencias en los factores de recombinación RAG1/RAG2, pueden originar, como mínimo, cuatro presentaciones clínicas distintas:
1. IDCS típica TB con intensa linfopenia.
2. Síndrome de Omenn.
3. IDCS T+B por injerto de células maternas.
4. Formas atípicas con presencia sustancial de células T, e incluso de células B y en ciertos casos con un número absoluto normal de linfocitos. En estos últimos casos la clínica puede ser muy atípica. Uno de estos casos fue diagnosticado en principio de artritis reumatoide (21).
Hoy sabemos que el Síndrome de Wiskott-Aldrich, con su tríada de eccema, trombocitopenia e infecciones de repetición, está causado por alteraciones en el mismo gen causante de la púrpura trombocitopénica ligada al cromosoma X (22). Son manifestaciones polares de una misma enfermedad, cuyo espectro clínico se mueve entre entre estos dos síndromes.
Las manifestaciones clínicas del síndrome linfoproliferativo ligado al cromosoma X (XLP), se resumen en la tabla IV. La clínica puede comenzar desde el año de vida hasta los 40 años y no existe una correlación entre el genotipo y el fenotipo, ya que se dan casos con distinto fenotipo en la misma familia (23).

Los defectos micobactericidas de los leucocitos, dan lugar a un cuadro de infecciones de repetición causados por micobacterias y salmonella. Tienen hasta el momento actual cuatro causas conocidas: alteraciones en una de las dos cadenas del receptor del IFN-g, la deficiencia de IL-12 y la deficiencia en el receptor de esta interleucina (24). Si bien no se puede hablar en sentido estricto de variantes, es importante conocer el defecto molecular que origina el síndrome, ya que la respuesta terapéutica al IFN-g depende de ella.
Quizás es la enfermedad granulomatosa crónica la IDP en la que se conoce desde hace más tiempo la existencia de formas variantes. Así las formas ligadas al sexo, suelen ser de peor pronóstico que las autosómicas recesivas, pero además existen pacientes en los que la enfermedad no se diagnostica hasta la pubertad, o inclusive en la edad adulta. Su clínica es más benigna y en algún caso hasta los 60 años no apareció ninguna infección mayor. Mutaciones que dan lugar a una formación residual de NADPH oxidasa, cercana al 5 % de lo normal, pueden permanecer asintomáticas durante años (25).
El síndrome autoinmune proliferativo, puede aparecer en heterozigotos. Siendo el FAS un trímero, alteraciones en una de las cadenas puede dar lugar a una unión defectuosa de la molécula con su ligando (9). Por otra parte, existen diferencias sustanciales entre unos y otros pacientes en cuanto a la gravedad del cuadro. Las mutaciones en el dominio intracelular de la proteína, se asocian a las manifestaciones más graves (10). Sin embargo dentro de una misma familia existen personas con la misma mutación y que no presentan patología mientras que otros sí (11). Por ello se supone que deben existir factores genéticos que protejan del fenotipo patológico.
Todavía podríamos dar muchos más ejemplos de la variabilidad clínica de estas enfermedades. Con frecuencia estas formas variantes son de difícil diagnóstico y es probable que algunos de los pacientes que tenemos clasificados como IDP no filiada, sean en realidad formas variantes de IDP conocidas.
Si bien las IDP son enfermedades con una incidencia baja y las formas variantes todavía mucho más infrecuentes, parece hoy en día necesario concienciarnos de que estas formas existen y deben ser diagnosticadas. Hay que tener presente que estos pacientes con enfermedades congénitas pero con clínica atípica, pueden pasar inadvertidas al pediatra y llegar con manifestaciones más floridas al internista o a otros especialistas de adultos. Otra lección que debemos aprender es que no siempre el fenotipo es sugerente de la enfermedad de base y que puede desviarse del considerado como característico, e incluso como patogénico de la enfermedad. Finalmente querría insistir en la necesidad de un diagnóstico precoz de estas enfermedades, tanto de las formas clásicas como de las variantes, ya que un diagnóstico temprano, es la mayor garantía del éxito terapéutico.
