



X-linked agammaglobulinemia (XLA) is characterized by absent or severely reduced B cells, low or undetectable immunoglobulin levels, and clinically by extracellular bacterial infections which mainly compromise the respiratory tract. We aimed to analyze the clinical, immunological and genetic characteristics of 22 male children with XLA.
MethodsTwenty-two children with XLA from 12 unrelated families were enrolled in this study. Clinical and demographic features of patients, serum immunoglobulin levels, percentage of B cells and BTK gene mutations were reviewed retrospectively.
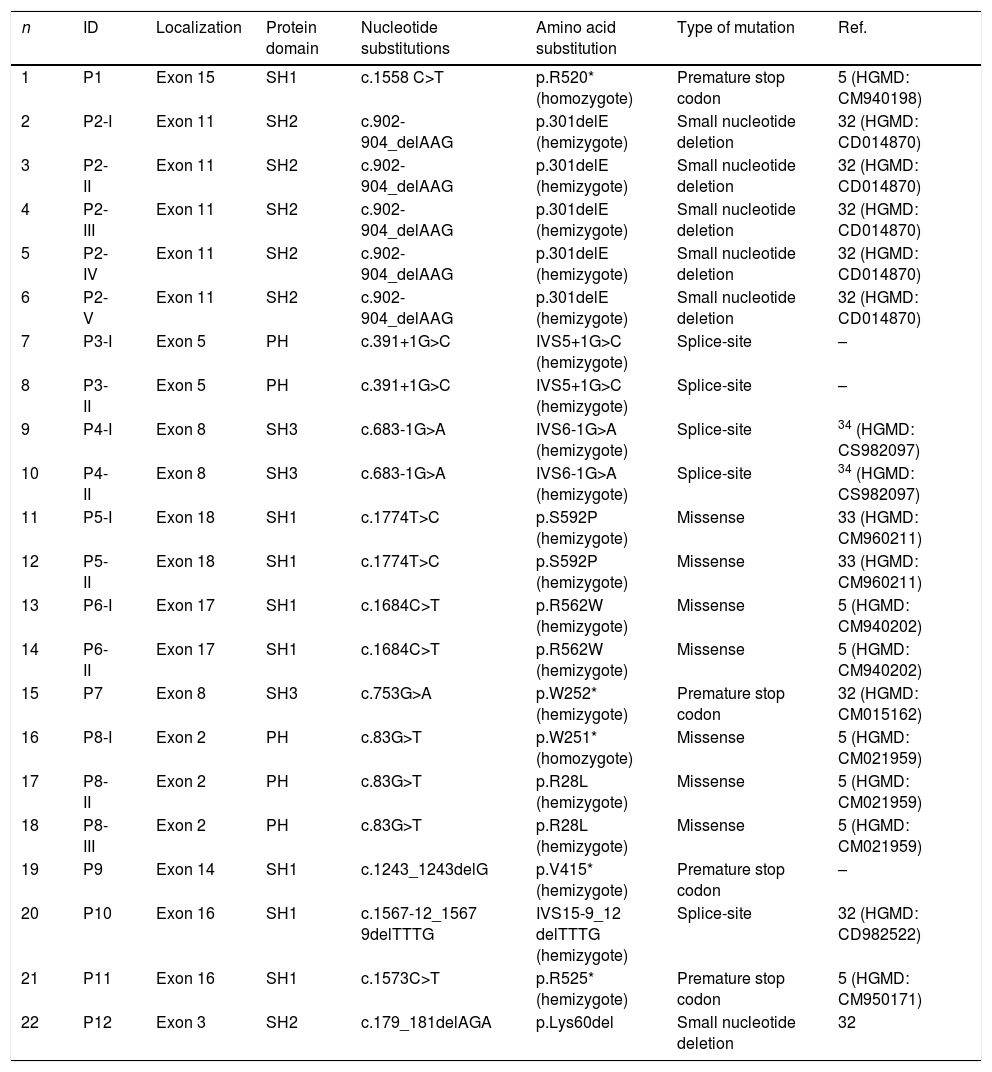
ResultsWe identified 12 different mutations in 22 patients from 12 unrelated families. The most frequent type of mutation was premature stop codon (33.3%). Ten mutations had been reported previously including three missense mutations (c.1774T>C, c.1684C>T, c.83G>T), three premature stop codons (c.1558C>T, c.1573C>T, c.753G>A), two splice-site (c.683-1G>A, c.1567-12_1567-9delTTTG) and two small nucleotide deletions (c.902-904_delAAG, c.179_181delAGA). Two novel mutations of the BTK gene were also presented and included one splice-site mutation (c.391+1G>C) and one premature stop codon mutation (c.1243_1243delG). Six out of 12 mutations of the BTK gene were located in the SH1 domain, two in the PH domain, two in the SH3 domain and two in the SH2 domain. Three patients had a history of severe infection before diagnosis. We did not identify any correlation between severity of clinical symptoms and the genotype.
ConclusionsOur results show that mutations in southeast Turkey could be different from those in the rest of the world and molecular genetic tests are an important tool for early confirmed diagnosis of XLA.
X-linked agammaglobulinemia (XLA) is characterized by a premature arrest in the development of B cells at the ProB cell stage due to deficiency of Bruton's tyrosine kinase (BTK) protein.1 Patients with XLA have a marked decrease in all isotypes of serum immunoglobulins (Ig) and almost a complete absence of peripheral B cells, defined by less than 2% B lymphocytes in the peripheral blood.2
The age of onset of the symptoms is usually after the 6th month of life which is in coincidence with the period where the maternal IgG fades.3 The patients suffer from recurrent bacterial infections associated with encapsulated, extracellular pyogenic pathogens such as Streptococcus pneumonae and Haemophilus influenzae.4
In order to decrease the frequency of infections and the risk of mortality, lifelong immunoglobulin replacement treatment is needed. The quality of life of XLA patients has significantly improved in the last two decades due to early diagnosis.3 Other indication types may be arthritis, osteomyelitis, sepsis, and more rare disorders such as hepatitis, vaccine-related polio, neutropenia, and autoimmune diseases.5
Although XLA was defined in 1952, the abnormal gene localized to Xq21.3-Xq22 was only detected in 1993 and termed as BTK.6 The human BTK gene encompasses 37.5kb comprising 19 exons and 18 of them encode protein. The protein consists of five different structural domains: pleckstrin homology (PH), Tec homology (TH), Src homology 3 (SH), SH2 homology, and kinase (SH1) domains. The mutations in BTK gene may affect any domains of the protein.7–9 The recent online BTK database contains 1375 entries collected from 1209 unrelated families with 742 unique mutations showing the majority of these mutations remotely. This information has been used to identify mutations in a series of patients with XLA who show various clinical manifestations and courses. Results from several studies’ reports suggest the presence of genotype–phenotype relationship.10,11 It is widely agreed that the specific mutation in BTK may be one of the factors that contributes to the severity of the disease.5,11 Nevertheless, it is important to consider that several factors such as age at diagnosis, the percentage of B cells, the concentration of serum immunoglobulins and polymorphic variants in Tec could be used to measure the clinical severity of XLA.10,12,13
The aim of our study was to evaluate the clinical profile and genotype/phenotype correlations in 22 Turkish male children with XLA from a single center.
Materials and methodsPatientsDuring 2008–2016, 22 male patients from 12 different families who were diagnosed with XLA were enrolled in this study. All of the patients were diagnosed at and followed up at the Department of Pediatric Allergy and Immunology, Çukurova University Medical Faculty. The diagnosis of XLA was made based on the diagnostic criteria of European Society for Immunodeficiencies (ESID) for primary immunodeficiency diseases. Definitive diagnosis of XLA was made if an BTK gene mutation was identified.14
The study was approved by the Ethical Committee of the institution and signed written informed consents were obtained from all parents.
Data collectionThe patients’ detailed clinical and laboratory data were retrospectively collected from the patients’ medical records, including clinical manifestations, age at onset and diagnosis, parental consanguinity, family history, type and severity of infections, immunological laboratory tests, treatments, and outcomes. The onset age was defined as the age when obvious infections initially occurred in the patients. The diagnosis age was taken as the age when immunodeficiency was identified in the patients.
Immunological analysisTotal serum IgG, IgA, and IgM levels were measured by nephelometry (BN ProSpec Systems, Date Behring Marburg GmbH, Marburg, Germany). Lymphocyte subsets from peripheral blood were numerated by means of flow cytometry with the following cluster of differentiation (CD) antigens: total T cells (CD3+), helper T cells (CD3+/CD4+), cytotoxic T cells (CD3+/CD8+), and B cells (CD19+) (Navios, Beckman Coulter, USA).
Genetic analysisGenetic testing to confirm the diagnosis of XLA was performed as part of genetic consultation with Medical Genetics Department of Çukurova University Faculty of Medicine. NGS was performed on MiSeq System, Illumina from leukocyte DNA from the cases of XLA patients. In silico analysis for novel mutations was carried out using SIFT, PolyPhen2 and MutationTaster.
Genotype–phenotype correlationMutations were classified into severe or less severe as previously described. Frameshift and premature stop codon leading to protein truncation were considered as severe mutations.10 The severity of the disease was assessed by considering the type of pre-diagnosis infections.
StatisticsStatistical analysis was performed using the statistical package SPSS software (Version 17.0, SPSS Inc., Chicago, IL, USA). All numerical data are expressed as mean values±SD or as proportions.
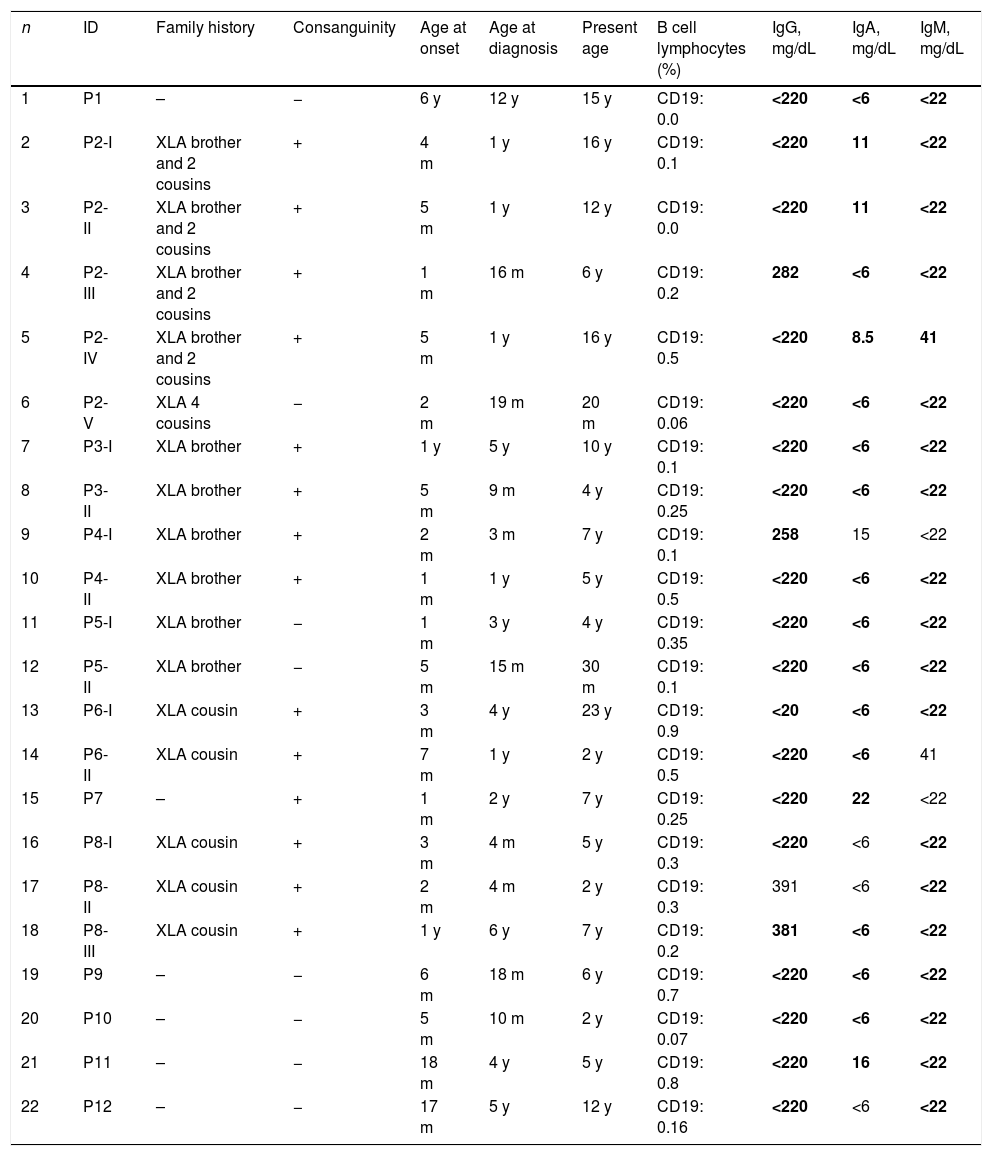
ResultsClinical characteristics22 male patients from 12 unrelated families were included in this study; all had proven mutations in BTK. Median age of patients during the study was 6 years (1.5–23). Median age at onset of symptoms was 5 months (1–72), median age at diagnosis was 15.5 months (3–144). The average delay in diagnosis was 20.95±20.78 (med: 11.5, 1–72) months. 86% of the patients were symptomatic in the first year of life and all of them were symptomatic by the age of 6 years. Fifteen of 22 XLA patients (68.1%) had a positive family history. Currently all of the patients are alive and receive regularly intravenous immunoglobulin (400–600mg/kg) treatment. Table 1 shows the characteristic features of these patients.
Characteristic features of 22 XLA patients.
| n | ID | Family history | Consanguinity | Age at onset | Age at diagnosis | Present age | B cell lymphocytes (%) | IgG, mg/dL | IgA, mg/dL | IgM, mg/dL |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | P1 | – | − | 6 y | 12 y | 15 y | CD19: 0.0 | <220 | <6 | <22 |
| 2 | P2-I | XLA brother and 2 cousins | + | 4 m | 1 y | 16 y | CD19: 0.1 | <220 | 11 | <22 |
| 3 | P2-II | XLA brother and 2 cousins | + | 5 m | 1 y | 12 y | CD19: 0.0 | <220 | 11 | <22 |
| 4 | P2-III | XLA brother and 2 cousins | + | 1 m | 16 m | 6 y | CD19: 0.2 | 282 | <6 | <22 |
| 5 | P2-IV | XLA brother and 2 cousins | + | 5 m | 1 y | 16 y | CD19: 0.5 | <220 | 8.5 | 41 |
| 6 | P2-V | XLA 4 cousins | − | 2 m | 19 m | 20 m | CD19: 0.06 | <220 | <6 | <22 |
| 7 | P3-I | XLA brother | + | 1 y | 5 y | 10 y | CD19: 0.1 | <220 | <6 | <22 |
| 8 | P3-II | XLA brother | + | 5 m | 9 m | 4 y | CD19: 0.25 | <220 | <6 | <22 |
| 9 | P4-I | XLA brother | + | 2 m | 3 m | 7 y | CD19: 0.1 | 258 | 15 | <22 |
| 10 | P4-II | XLA brother | + | 1 m | 1 y | 5 y | CD19: 0.5 | <220 | <6 | <22 |
| 11 | P5-I | XLA brother | − | 1 m | 3 y | 4 y | CD19: 0.35 | <220 | <6 | <22 |
| 12 | P5-II | XLA brother | − | 5 m | 15 m | 30 m | CD19: 0.1 | <220 | <6 | <22 |
| 13 | P6-I | XLA cousin | + | 3 m | 4 y | 23 y | CD19: 0.9 | <20 | <6 | <22 |
| 14 | P6-II | XLA cousin | + | 7 m | 1 y | 2 y | CD19: 0.5 | <220 | <6 | 41 |
| 15 | P7 | – | + | 1 m | 2 y | 7 y | CD19: 0.25 | <220 | 22 | <22 |
| 16 | P8-I | XLA cousin | + | 3 m | 4 m | 5 y | CD19: 0.3 | <220 | <6 | <22 |
| 17 | P8-II | XLA cousin | + | 2 m | 4 m | 2 y | CD19: 0.3 | 391 | <6 | <22 |
| 18 | P8-III | XLA cousin | + | 1 y | 6 y | 7 y | CD19: 0.2 | 381 | <6 | <22 |
| 19 | P9 | – | − | 6 m | 18 m | 6 y | CD19: 0.7 | <220 | <6 | <22 |
| 20 | P10 | – | − | 5 m | 10 m | 2 y | CD19: 0.07 | <220 | <6 | <22 |
| 21 | P11 | – | − | 18 m | 4 y | 5 y | CD19: 0.8 | <220 | 16 | <22 |
| 22 | P12 | – | − | 17 m | 5 y | 12 y | CD19: 0.16 | <220 | <6 | <22 |
m=month; y=year.
Low immunoglobulin levels are marked in bold according to same age healthy children.31
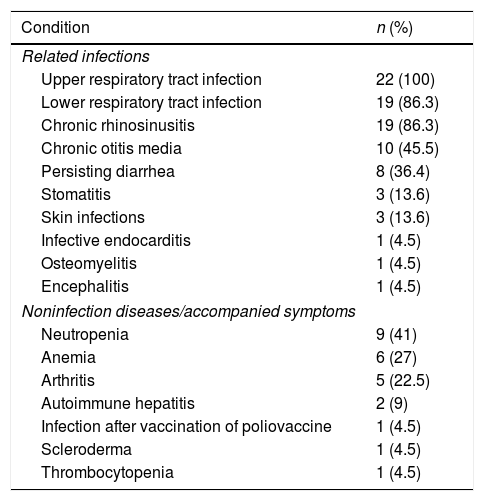
Clinical presentations of infection were identified in patients before XLA was diagnosed, all of those were upper respiratory tract infections. Lower tract infection was found in 19 cases (86.3%), whereas otitis media in 10 cases (45.5%) and chronic diarrhea in eight cases (36.4%). Three patients suffered from severe infections, such as osteomyelitis (P7), infective endocarditis (P1) and encephalitis (P12). As illustrated in Table 2, six patients had other complications such as arthritis (four cases), autoimmune hepatitis (two cases), scleroderma (one case), and infection after polio vaccine (one case). The clinical manifestations in XLA patients are shown in Table 2.
Frequency of related infections and accompanied symptoms in XLA patients.
| Condition | n (%) |
|---|---|
| Related infections | |
| Upper respiratory tract infection | 22 (100) |
| Lower respiratory tract infection | 19 (86.3) |
| Chronic rhinosinusitis | 19 (86.3) |
| Chronic otitis media | 10 (45.5) |
| Persisting diarrhea | 8 (36.4) |
| Stomatitis | 3 (13.6) |
| Skin infections | 3 (13.6) |
| Infective endocarditis | 1 (4.5) |
| Osteomyelitis | 1 (4.5) |
| Encephalitis | 1 (4.5) |
| Noninfection diseases/accompanied symptoms | |
| Neutropenia | 9 (41) |
| Anemia | 6 (27) |
| Arthritis | 5 (22.5) |
| Autoimmune hepatitis | 2 (9) |
| Infection after vaccination of poliovaccine | 1 (4.5) |
| Scleroderma | 1 (4.5) |
| Thrombocytopenia | 1 (4.5) |
IgG, IgA, and IgM levels and percentage of B cells are shown in Table 1. Serum IgG was <220mg/dL in 18 patients at diagnosis. Patients 17 had normal serum IgG levels (391mg/dL), but his IgA and IgM levels were extremely low. The percentage of B cells of these patients was 0.3%. The percentage of B cells in all patients was lower than 1%.
BTK mutation analysisMutation analysis of the BTK gene was performed in 22 patients in 12 different families, including premature stop codon in four families, missense mutation in three families, splice-site mutations in three and small nucleotide deletion in two families. Of the 12 BTK gene mutations, 10 had been reported previously including three missense mutations (c.1774T>C, c.1684C>T, c.83G>T), three premature stop codon (c.1558 C>T, c.753G>A, c.1573C>T), two splice-site (c.683-1G>A, c.1567-12_1567-9delTTTG), and two small nucleotide deletion (c.902-904_delAAG, c.179_181delAGA). Two novel mutations of the BTK gene were also presented and included one splice-site mutation (c.391+1G>C) and one premature stop codon mutation (c.1243_1243delG) (Patients 3 I-II and 9, respectively) (Fig. 1a, b). Six out of 12 mutations of the BTK gene were located in the SH1 domain, two in the PH domain, two in the SH3 domain and two in the SH2 domain (Table 3). Three patients had a history of severe infection prior to diagnosis and premature stop codon mutation was present in the BTK gene in two patients among them.
 NGS data electropherogram tracing of the exon 5 in BTK gene showing the novel homozygous mutation c. Patient Number 9: (b) NGS data electropherogram tracing of the exon 14 in BTK gene showing the novel homozygous mutation c.")
Sequence view of novel mutations in BTK gene of the cases. Parents scanned for the same mutation in order to confirm the data collected, and heterozygous mutations were detected in the same position and defined as a carrier.
Patient Number 3-I and 3-II: (a) NGS data electropherogram tracing of the exon 5 in BTK gene showing the novel homozygous mutation c.
Patient Number 9: (b) NGS data electropherogram tracing of the exon 14 in BTK gene showing the novel homozygous mutation c.
Btk mutation analysis in patients with XLA in this study.
| n | ID | Localization | Protein domain | Nucleotide substitutions | Amino acid substitution | Type of mutation | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | P1 | Exon 15 | SH1 | c.1558 C>T | p.R520* (homozygote) | Premature stop codon | 5 (HGMD: CM940198) |
| 2 | P2-I | Exon 11 | SH2 | c.902-904_delAAG | p.301delE (hemizygote) | Small nucleotide deletion | 32 (HGMD: CD014870) |
| 3 | P2-II | Exon 11 | SH2 | c.902-904_delAAG | p.301delE (hemizygote) | Small nucleotide deletion | 32 (HGMD: CD014870) |
| 4 | P2-III | Exon 11 | SH2 | c.902-904_delAAG | p.301delE (hemizygote) | Small nucleotide deletion | 32 (HGMD: CD014870) |
| 5 | P2-IV | Exon 11 | SH2 | c.902-904_delAAG | p.301delE (hemizygote) | Small nucleotide deletion | 32 (HGMD: CD014870) |
| 6 | P2-V | Exon 11 | SH2 | c.902-904_delAAG | p.301delE (hemizygote) | Small nucleotide deletion | 32 (HGMD: CD014870) |
| 7 | P3-I | Exon 5 | PH | c.391+1G>C | IVS5+1G>C (hemizygote) | Splice-site | – |
| 8 | P3-II | Exon 5 | PH | c.391+1G>C | IVS5+1G>C (hemizygote) | Splice-site | – |
| 9 | P4-I | Exon 8 | SH3 | c.683-1G>A | IVS6-1G>A (hemizygote) | Splice-site | 34 (HGMD: CS982097) |
| 10 | P4-II | Exon 8 | SH3 | c.683-1G>A | IVS6-1G>A (hemizygote) | Splice-site | 34 (HGMD: CS982097) |
| 11 | P5-I | Exon 18 | SH1 | c.1774T>C | p.S592P (hemizygote) | Missense | 33 (HGMD: CM960211) |
| 12 | P5-II | Exon 18 | SH1 | c.1774T>C | p.S592P (hemizygote) | Missense | 33 (HGMD: CM960211) |
| 13 | P6-I | Exon 17 | SH1 | c.1684C>T | p.R562W (hemizygote) | Missense | 5 (HGMD: CM940202) |
| 14 | P6-II | Exon 17 | SH1 | c.1684C>T | p.R562W (hemizygote) | Missense | 5 (HGMD: CM940202) |
| 15 | P7 | Exon 8 | SH3 | c.753G>A | p.W252* (hemizygote) | Premature stop codon | 32 (HGMD: CM015162) |
| 16 | P8-I | Exon 2 | PH | c.83G>T | p.W251* (homozygote) | Missense | 5 (HGMD: CM021959) |
| 17 | P8-II | Exon 2 | PH | c.83G>T | p.R28L (hemizygote) | Missense | 5 (HGMD: CM021959) |
| 18 | P8-III | Exon 2 | PH | c.83G>T | p.R28L (hemizygote) | Missense | 5 (HGMD: CM021959) |
| 19 | P9 | Exon 14 | SH1 | c.1243_1243delG | p.V415* (hemizygote) | Premature stop codon | – |
| 20 | P10 | Exon 16 | SH1 | c.1567-12_1567 9delTTTG | IVS15-9_12 delTTTG (hemizygote) | Splice-site | 32 (HGMD: CD982522) |
| 21 | P11 | Exon 16 | SH1 | c.1573C>T | p.R525* (hemizygote) | Premature stop codon | 5 (HGMD: CM950171) |
| 22 | P12 | Exon 3 | SH2 | c.179_181delAGA | p.Lys60del | Small nucleotide deletion | 32 |
Novel mutations were identified in patients 3 and 9.
In the present study, we determined 22 XLA patients who had 12 different mutations in BTK. Novel mutations were identified in two of 12 and 10 previously described mutations. The most frequent type of mutation was premature stop codon (33.3%). All of the patients were admitted due to infections, and upper respiratory tract infection was the most common symptom.
We found that the spectrum of mutations had no correlation with the previously reported study group of BTK mutation in other countries.15–17 However, similar mutation spectrum has been detected in the BTK gene in the previously reported analyses from our country.17 Stop codon protein mutations were the most common followed by missense, splice site, small nucleotide deletion in our study. These data indicate that BTK mutation profiles in Turkey could be different from those in the rest of the world, necessitating large studies on different ethnic populations. On the other hand, six out of 12 BTK mutations (50%) were located on the SH1 domain in our study, consistent with other studies’ reports in the literature.17–19 This might be explained by the extent of the SH1 domain.19
Infection is the initial manifestation of XLA patients. In our study, all of the patients had a history of infection before the diagnosis. Clinical symptoms identified in the patients showed in this study include those previously observed in XLA, such as respiratory tract infections.20–23 Chronic rhinosinusitis (86.3%) was also very common, whereas skin infections (13.6%) and stomatitis (13.6%) were less frequent. When assessed in terms of severe infection, three patients had osteomyelitis (P7), infective endocarditis (P1) and encephalitis (P6-I). Two of the patients (P1,7) among them had premature stop codon mutation. On the other hand, the other two patients (P9,11) with premature stop codon mutation did not have severe infection histories. Our results suggest that the correlation between the specific mutation in BTK and the severity of the disease alone may not be sufficient to predict clinical progress in a patient with XLA based on the mutation.
The frequency of arthritis in XLA patients has been reported to be variable, ranging from 7% to 35% according to the ethnic background.15,24,25 In our study, the frequency of arthritis is similar to previous reports, but reactive arthritis is found in all of them. None of the patients proceeded to chronic arthritis and responded well to the non-steroidal anti-inflammatory drugs. Additionally, we analyzed the BTK mutation status and the domains affected for each arthritic patient. It was reported that missense mutations were more frequent in patients who had developed arthritis.15 Interestingly, in our cases with arthritis, whilst two of them had small nucleotide deletions on SH2 domains, one had a splice site deletion on SH1 domain. In the other two, one had mutation on splice site of PH domain and the other one had missense mutation on SH1 domain.
Arthritis and primary immune diseases including XLA may be related, although the mechanisms are not fully elucidated. Furthermore, BTK is important for B cell development and function, and there is increasing evidence that it is associated with TLR-mediated activation of innate immune cells such as neutrophils and monocytes. It is known that these cells produce pro-inflammatory cytokine and reactive oxygen radicals. BTK is a negative regulator of Toll-like receptors (TLRs) in innate cell activation, and this function is mediated by MyD88-adaptor-like (MAL) interactions and disruption. The SH1 and PH domains interact with TLRs, which is an important signaling molecule related to cell activation through MAL.26 This condition is considered to contribute to the susceptibility to inflammatory conditions.27 Virtually all patients had infections such as chronic sinusitis, otitis and/or diarrhea in our study. Our results also suggest that the development of arthritis might be due to exaggerated inflammatory responses to circulating microbial products.
X-linked agammaglobulinemia might be associated with some autoimmune diseases.27–29 One of our patients was diagnosed with scleroderma. The diagnosis of scleroderma was made based on physical examination and skin biopsy. Localized scleroderma (LoS) is a chronic connective tissue disease of unknown etiology and characterized by skin thickening with inflammation and fibrosis. LoS may accompany other autoimmune and collagen tissue diseases.30 Our patient had monoarthritis and hepatitis in addition to LoS.
In conclusion, we have identified our mutation profile to be different from those in the rest of the world. In the literature, the presence of missense mutations are the hot spots, where one is in the SH1 and the other is in the PH domain. However, in our patient series, the most common hot spot regions were distinctive with 50% of SH1 domain, 16.6% mutations on PH domain and the other in SH2 (16.6%) and SH3 (16.6%) domains. In addition, the absence of a strong genotype/phenotype relation in our patients indicates the presence of other factors that may be effective in the course of the disease.
Ethical disclosuresConfidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and\ or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that no experiments were performed on humans or animals fort this investigation.
Conflict of interestThe authors declare that they have no conflict of interest.
