


In recent years our concept of the non-specific nature of innate immunity has changed following the identification of a network of germline-encoded receptors that recognise with substantial specificity molecular motifs of microorganisms and many other cues produced during tissue injury. Stimulation of these innate sensors by their specific ligands triggers signalling pathways that result in the activation of innate effector mechanisms as well as the priming of naive lymphocytes for the type of response that must be induced. These events culminate in the generation of an immune response appropriately adapted to the damage that has occurred. These new insights into innate immunity herald an entirely new era in the understanding of the molecular events that initiate and drive a host-protective response, changing many concepts about susceptibility to infections and providing greater insight into the underlying inflammatory pathology of other diseases. Targeted manipulation of innate immunity has enormous potential for the development of new vaccines and innovative therapies for the treatment of diseases such as infections, cancer, allergy, autoimmunity and autoinflammatory diseases. This article provides an overview of current trends in the field of innate immunity and its role in the control of infection and disease.
One of the distinctive features of the host defence system in mammals is the presence of specialized cells that detect pathogens and activate effector mechanisms to control and destroy invasive microorganims. It is comprised of two branches: innate and acquired immunity. Cells involved in innate immunity contribute to the control and eradication of infectious agents by activating evolutionarily conserved effector mechanisms of defence and clearance. This response is characterized by its rapid induction, as is required in an emergency situation, and is responsible for host defence during the initial days of infection, but it does not generate lasting protective immunity. The adaptive immune response takes longer to develop but specifically recognizes microorganisms to activate a response that amplifies the effector mechanisms involved in innate immunity and in this way destroy a particular pathogen more efficiently. This response allows the organism to react quickly to subsequent reinfections.
The belief that a highly specialized adaptive immunity is essential for the effective protection of mammals against pathogens has for many years focused scientific interest on this final part of the immune response. In contrast, innate immunity has been regarded as a primitive ancestor of adaptive immunity that provides only a non-specific defensive response characterized by phagocytosis of microorganisms, foreign substances or detritus, and its contribution to immune protection has been underestimated. In recent years, various studies of immunity in plants and insects have identified phylogenetically conserved mechanisms of surveillance and defence that offer effective protection against deadly pathogens, despite the absence of an adaptive immunity in those organisms. In these systems, pathogens are attacked with burst of superoxide, hydrogen peroxide, nitric oxide or toxic antimicrobial metabolites. However, these organisms have also developed strategies to detect pathogens and selectively activate an immune response. These mechanisms are mediated by products encoded in the host genome that have been conserved during evolution and are presently found throughout the animal and plant kingdoms. In mammals, signals generated by innate immunity also participate in the polarization and modulation of adaptive immunity. These new concepts reveal innate immunity to be more specific than previously thought and suggest that it plays a more prominent role in host protection.
INNATE IMMUNITY IDENTIFIES PATHOGENS AND SENSES DAMAGECharles Janeway1,2 was the first to explore the “road not taken” of the innate immune response, and in the late 1980s he established new paradigms for innate immunity. He had the idea that infectious agents activate naive T cells via their effects on the innate immune system and proposed that cells involved in innate immunity recognize pathogens through germline-encoded receptors, each harbouring a fixed specificity. He coined the term “pattern recognition receptors” or PRRs to refer to such molecules. PPRs have been selected by innate immunity throughout evolution to detect invariant molecular constituents expressed by a wide range of different microorganisms2,3. These molecular constituents, which are not expressed in the host, were termed “pathogen-associated molecular patterns” or PAMPs. PAMPs are essential for survival of the microorganism, since mutations affecting the genes that encode them will lead to non-viable organisms. Thus, many of them are highly conserved molecules that are present in thousands of microorganisms. This host immune strategy helps to avoid mutants escaping immune surveillance and allows the immune system to detect many classes of microorganisms through the use of a relatively limited repertoire of receptors. The fact that PAMPs are produced only by the invading microbes and are not expressed in the host allows innate immunity to discriminate between “infectious non-self” and “non-infectious self”.4,5
At the end of the 1990s, several studies in Drosophila revealed that the Toll receptor, a protein involved in embryonic development of the fly and which shows homology to the interleukin (IL) 1β receptor, is a key mediator of innate immunity in that model system.6,7 At the time that these results were published, Charles Janeway's group had cloned the mammalian homologue of Toll and shown that a dominant active form of the protein could activate NFκB and several NFκB-dependent inflammatory genes, and induce the expression of the co-stimulatory molecules CD80 and CD86.8 These results revealed that costimulatory signals required for the activation of naive T cells are generated via PRR-PAMP interactions, demonstrating that innate receptors can induce signals that activate both an innate and an adaptive immune response. Later, this receptor was named Toll-like receptor (TLR) 4,9 and was identified as a lipopolysaccharide sensor involved in susceptibility to infection by gram-negative bacteria.10,11 Since then, 13 different TLRs have been identified in mouse and analysis of the human genome database has revealed the existence of 11 TLR homologs in humans.9,12
Janeway's work explained how the immune system evolved to discriminate infectious non-self from non-infectious self; many non-infectious factors, however, such as traumatic injury, transplantation, or exposure to a long list of chemical and physical insults, cause tissue damage that must be controlled and repaired by immune cells in the absence of infectious non-self signals. In the early 1990s, Polly Matzinger, in another view of innate immunity, postulated the “danger model”, which proposed that cells of the innate immune system detect those substances that are dangerous rather than simply foreign.13–15 She stated that under stressful conditions, dead or damaged cells release endogenous molecules that should not normally be present outside the cell or in certain intracellular locations, and that these serve as early warning signals to activate primary and secondary immune responses. The term “alarmin” was proposed by Oppenheim to refer to endogenous stress molecules that signal tissue and cell damage.16,17 Alarmins are rapidly released by dead cells but not by apoptotic cells in response to infection or tissue injury. They have both chemotactic and activating effects on innate and adaptive immune responses, possess in vivo immune-enhancing activity, and promote reconstruction of the damaged tissue. Thus, this subset of mediators alerts host defences and promotes an immune response both after tissue injury and infection. Mediators with alarmin activity include a long list of molecules, among them defensins, cathelicidin, eosinophil-derived neurotoxin, high mobility group box protein 1, protein S100, ATP and uric acid.18 Endogenous alarmins and exogenous PAMPs are currently considered subgroups of a larger set, the damage-associated molecular patterns or DAMPs.14,19 Recently it has been proposed that DAMPs might be part of an evolutionarily ancient warning system in which the hydrophobic portions of biological molecules, when exposed, act as universal DAMPs to initiate repair, remodelling, and immune response.20
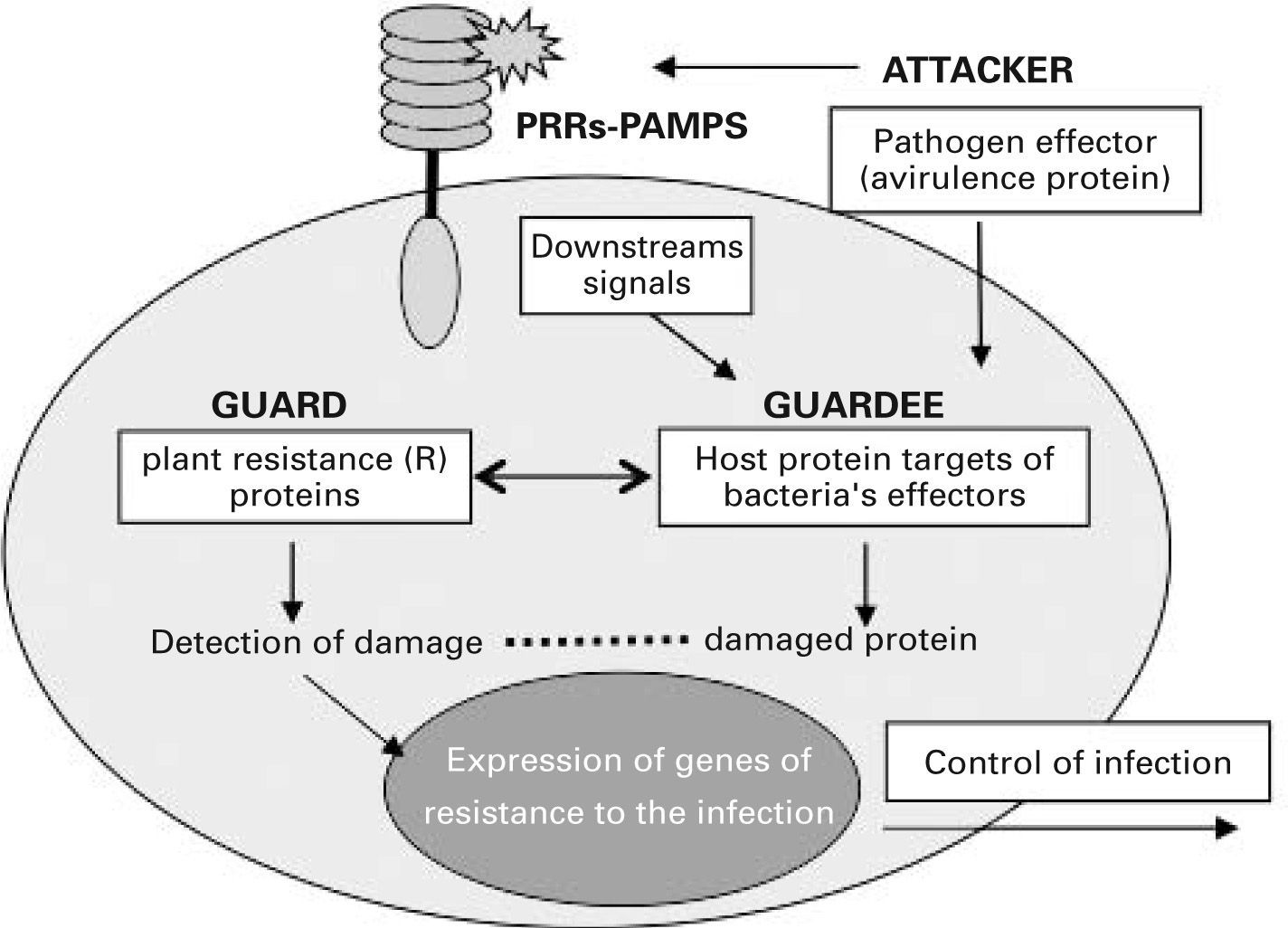
Many of the mechanisms used by the innate immune system in animals show surprising parallels with those of immunity in plants, and this has highlighted common research goals for immunologists working with both systems.21 Unlike mammals, plants lack motile cells involved in host defence and each of their somatic cells must have the capacity to perceive attacks by pathogens and alert other cells to the infection. Plants protect against pathogens via a two-branched innate immune system.22 One branch uses transmembrane PRRs that recognize and respond to PAMPs through a signalling pathway that is reminiscent of the mammalian one. However, during the coevolution of host-microbe interactions, pathogens acquired the ability to deliver effector factors to the plant cell which suppress PAMP-triggered immunity, thus allowing pathogen growth. The other branch of innate immunity in plants senses these pathogen effector factors through intracellular PRRs, such as the plant resistance proteins (R proteins). These receptors are proteins with leucine-rich repeat (LRR) motifs and nucleotide binding oligomerization (NOD) domains and associate in multiprotein complexes that detect the presence of pathogens and induce a strong resistance response to the infection.23 From the plant research community emerged in 1998 the “guard hypothesis”,24,25 which postulated that plant resistance proteins act as guards for cellular machinery by protecting those components of the host cells that are targets of bacterial effectors. It is thought that these proteins somehow sense the damaged status of the effector targets, ra ther than the effectors themselves, and activate resistance (see fig. 1). According to this theory, perturbation of the host target generates a “pathogen-induced modified self” which activates the plant resistance proteins, triggering resistance to the infection. This concept is analogous to the immune recognition of “modified self” proposed for mammals in the danger model.26 These proposals support the idea that host cells do not search for microbial products but rather protect themselves such that when a physiological disturbance occurs an immune response is initiated. These new concepts regarding innate immune surveillance represent a much larger step toward understanding how immunity functions to protect the host, since they suggest that innate immunity has developed strategies to differentiate between normal and damaged or diseased cells.
 into the host cell. These effectors alter the function of proteins involved in downstream signalling from membrane pattern recognition receptors (PRRs). Intracellular PRRs (guards) somehow sense pathogeninduced damage of their “guardee” protein and trigger a resistance response against the infection that renders the pathogen avirulent. PAMPs, pathogenassociated molecular patterns.")
The guard hypothesis. Invading plant pathogens deliver effector molecules (termed avirulence proteins) into the host cell. These effectors alter the function of proteins involved in downstream signalling from membrane pattern recognition receptors (PRRs). Intracellular PRRs (guards) somehow sense pathogeninduced damage of their “guardee” protein and trigger a resistance response against the infection that renders the pathogen avirulent. PAMPs, pathogenassociated molecular patterns.
In the last 8years, a family of at least 22 mammalian genes that encode intracellular receptor homologues to plant resistance proteins has been identified.27,28 These receptors possess NOD and LRR domains, and form protein complexes that respond to PAMPs and danger signals, playing an important role in the human immune response. On the basis of their structure and function, this new class of intracellular PRRs has been named NOD-like receptors or NLRs.
THE NETWORK OF INNATE RECEPTORS THAT SENSE DAMAGE AND ALERT THE IMMUNE SYSTEMIt seems clear that the strategy of innate immunity to identify and specifically respond to the presence of a broad class of pathogens and injuries is based on the existence of a set of PRRs in all cells of the same cell type that can discriminate between different DAMPs5. The combined activation of these different receptors by their specific ligands triggers signalling pathways that can be complementary, synergistic or antagonistic.29–31 This functional cooperation and complementation between receptors, in association with some accessory molecules, results in the activation of specialized effector mechanisms to destroy a given pathogen and in the generation of signals that provide information to the adaptive immune system about the origin of the antigen and the type of adaptive response to be induced.32–35 In addition, several cellular strategies have evolved to negatively regulate immune activation induced through PRRs in order to prevent inappropriate or overactive responses. Negative regulators act at multiple levels within the PRR signalling cascades or elicit negative-feedback mechanisms that synchronize the positive activation and negative regulation of signal transduction.36–39 These various elements together reveal a tightly controlled innate receptor network that monitors changes in tissue homeostasis to alert and drive both innate and adaptive immunity.
PRRs CLASSIFICATIONMammalian PRRs can be functionally classified into three main groups: humoral proteins, endocytic receptors, and signalling receptors.4,29 Humoral proteins identify, bind and opsonize the pathogen to neutralize and clear it through activation of the complement and phagocytic systems. These PRRs include mannose-binding lectins, C-reactive proteins and collectins. Endocytic receptors, including some C-type lectins and scavenger receptors, directly support the capture of pathogens or damaged cells and promote their internalisation and transport to the lysosomal compartment to be degraded. In addition to their role in the effector response, both groups have a critical role in activating antigen presentation. Signalling receptors that act as primary sensors of pathogens and damage trigger intracellular signalling cascades that result in the upregulation of immune response genes which are critical for the induction of both effector and adaptive responses. This last group of receptors can be located on the plasma membrane, in different internal compartments, or in membranes from intracellular vesicles, or they can be cytosolic proteins, and their expression is not restricted to cells of the immune system. Three families of signalling PRRs have been identified: TLRs, NLRs and retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs)40.
Receptors for alarmins include the IL-1 receptor, RAGE (a multiligand receptor binding advanced glycation end products) and RPTP (receptor-type tyrosine phosphatase) β/ζ; however, there is increasing evidence that in order to signal alarm most endogenous molecules produced by injured tissues bind the same TLRs and NLRs as microorganisms and evoke similar responses.41,42 That suggests that PRRs and receptors for alarmins synergistically reinforce each other to evoke an immune response, but it could also mean that both PAMPs and endogenous alarm signals belong to a common set of signals that are nearly as ancient as life itself, as recently proposed by Polly Matzinger.20
There is also a group of transmembrane receptors that sense pathogens and directly or indirectly function as PRRs and cooperate with them.43,44 These receptors include members of the TREM (triggering receptors expressed on myeloid cells) proteins. Among them, TREM-1 synergistically amplifies signals mediated by TLRs and NLRs to upregulate production of proinflammatory cytokines. In addition to roles in adhesion and endocytosis, many receptors of the C-type lectin family, particularly dectin-1 and DC-SIGN, can also mediate cell signalling for effector responses and, in particular, cooperate with TLRs to enhance or inhibit them. Various members of the family of Siglecs (sialic acid-binding immunoglobulin-like lectins) possess a conserved immunoreceptor tyrosine-based inhibitory motif (ITIM) and an ITIM-like motif in the cytoplasmic tail, indicating that they have roles in modulating cell function. It is thought that these receptors promote cell-cell interactions and regulate the functions of cells in the innate and adaptive immune systems through glycan recognition.44
TLRS IN INNATE AND ADAPTIVE IMMUNITYTLRs are a family of PRRs capable of detecting a long list of DAMPs and inducing the expression of proinflamatory genes and costimulatory molecules. All TLRs possess an ectodomain composed of an LRR motif involved in recognition of PAMPs and a cytoplasmic signalling domain that shows a remarkable homology to the cytoplasmic region of the IL-1 receptor, called the Toll IL-1 receptor (or TIR) domain, which is required for downstream signalling.12 Stimulation of these receptors by their specific ligands results in the acquisition of specialized effector functions of innate cells but TLRs expressed on dendritic cells (DC) have been shown to be critical for the generation of signals that polarize the adaptive immune response,34,45,46 placing these receptors at the interface between innate and adaptive immunity. The importance of TLRs in the outcome of adaptive responses is supported by analysis of genetic polymorphisms, mutations and experimental models that reveal associations with susceptibility to infection and with immune disorders such as allergy,47–49 autoimmunity50–52 and cancer.53,54
Studies to identify potential sites of TLR action have revealed that most of the tissues express mRNA at least for one TLR and several express all of them, although changes in the expression of TLR mRNA may be induced by purified PAMPs and cytokines.55,56 Notable differences have been observed between human and mouse TLR expression, but in both species the largest repertoire has been detected in cells and tissues involved in innate immunity.55,57 At the cellular level, TLRs are differentially distributed and their location correlates with the nature of their ligands. Those located in the plasma membrane recognize external ligands at the cell surface, whereas TLRs located in membranes from intracellular vesicles such as endosomes only detect ligands present in the lumen of those structures, mainly viral nucleic acids.12,58–60
A pathogen gaining entry to the host or release of alarm signals as a result of injury is sensed by sentinel cells through TLRs and the cells quickly respond by activating a cascade of biochemical events that initiates inflammation.61 Although TLRs expressed in resident macrophages, mast cells and endothelial cells mainly perform this function, TLRs constitutively expressed by other non-immune cells such as smooth muscle cells or fibroblasts are also involved.62 In inflamed tissues, there is a marked upregulation of TLR expression in these somatic cells, allowing recognition of both PAMPs and endogenous agonists generated to reinforce and amplify the inflammatory response.63 Indirectly, TLRs expressed in somatic cells also mediate the release of molecules that prime DCs to acquire a determined function.64 When expressed on epithelial barriers, TLRs play an essential role in maintaining tolerance to commensal microorganisms.54
One of the most intriguing recent observations is that TLRs are also expressed on T and B cells and that they respond to their ligands by modulating and cooperating with adaptive immunity in different steps.65–68 Stimuli mediated by PRRs expressed on naive B cells optimise the sequential integration of signals mediated by antigen presentation to specific helper T (Th) cells through immune synapses.65,66 TLRs are also expressed on different subsets of T lymphocytes and their respective ligands can directly modulate T cell function. With few exceptions, it appears that TLR signalling modulates T cell responses triggered by TCR stimulation rather than inducing a direct cellular response.68
NLRS AND RLRS: INTRACELLULAR SENSORS OF INFECTION AND DAMAGENLRs are a family of intracellular proteins with a tripartite modular structure that contain a central nucleotide-binding oligomerization domain, a C-terminal LRR, and an N-terminal effector-binding domain that shares structural similarity with a subclass of plant disease resistance genes.27,69 Various studies have found evidence that NLR activation occurs through a mechanism similar to that described for the formation of the apoptosome, although their structural characteristics remain to be elucidated.69 It has been proposed that NLR proteins are present in the cytoplasm in a monomeric auto-repressed form and that the presence of bacterial products in the cytoplasm directly or indirectly induces a conformational rearrangement of the NLR molecule that leads to auto-association to form a complex protein structure in which the receptor acts as a central scaffold protein to recruit the downstream effector molecules and integrate cellular immune signals.69 Several NLRs (NALPs and IPAF subfamilies) form multi-protein complexes termed inflammasomes, which engage inactive proforms of the enzyme Caspase-1. This induced proximity of two inflammatory pro-caspases initiates their autoprocessing, which leads to enzyme activation and the subsequent cleavage of pro-IL-1β, pro-IL-18 and probably pro-IL-33 to generate mature and active forms.70,71
Under many conditions of cellular damage or under physical or psychological stress, ATP is released from cells without cell lysis, and this is considered as a danger signal that induces the formation of a NALP inflammasome and Caspase-1 activation.72 Recently it has been reported that monosodium urate (MSU) and calcium pyrophosphate dihydrate crystals, the causative agent of gout and pseudogout inflammatory diseases, engage the Caspase-1-activating NALP inflammasome and induce the release of inflammatory cytokines.73 It was proposed that uric acid could nucleate to form MSU and act as a danger signal that activates Caspase-1, further supporting the role of NLRs as intracellular danger sensors.
Other members of the NLR family (the NOD subfamily) also form complex protein structures termed nodosomes, which recruit the downstream effector molecules initiating a signalling pathway that leads to the activation f NFκB and to the expression of inflammatory genes.74
Genetic alterations in NLRs are linked to autoinflammatory syndromes such as familial cold autoinflammatory syndrome, Muckle-Wells syndrome and chronic infantile neurological cutaneous and articular syndrome/neonatal onset multisystemic inflammatory disease.75,76 Other mutations affecting genes that code for proteins implicated in the regulation of inflammasome assembly and Caspase-1 activation are responsible for other very similar autoinflammatory diseases. Such is the case for familial Mediterranean fever,77 pyogenic arthritis, pyoderma gangrenosum, and acne syndrome,78 or hyperimmunoglobulinemia D syndrome.79 Mutations that affect nodosome formation have been associated with other autoinflammatory diseases such as Blau syndrome and early-onset sarcoidosis, and with susceptibility to Crohn's disease.76,80
The RLR family includes proteins encoded by RIG-I, the melanoma differentiation-associated gene MDA5 and the laboratory of genetics and physiology 2 gene LGP2, which detect intracellular double-stranded RNA and activate certain interferon-regulated factor family members. These proteins have been considered as cytosolic sensors of viruses that protect all virally-infected cells. It has recently been reported that RLRs are expressed ubiquitously, suggesting that these sensors represent the foremost antiviral defence system in most organs.81,82 It is likely that TLRs and RLRs function together to provide omnipresent anti-viral protection, with anti-viral TLRs being more restricted to DCs. These receptors have been implicated in autoimmunity and recently it has been suggested that they might be involved in mouse development, independently of their role as virus sensors.83
DENDRITIC CELLS: THE MAIN LINK BETWEEN INNATE AND ADAPTIVE IMMUNITYThe specificity of adaptive immunity in identifying pathogens is based on the existence of a varied repertoire of antigen receptors that are generated by somatic mechanisms during the ontogeny of each individual organism. These receptors are clonally distributed on T and B cells and possess random specificities. Unlike cells involved in innate immunity, specific lymphocytes are not pre-programmed for a particular effector response and acquisition of a specialized function requires cross-talk between naive lymphocytes and antigen-presenting cells (APCs). This communication is mainly mediated by signals generated through PRR-PAMP interactions, which alert the innate immune system to the presence of microbial infection or tissue damage to initiate a rapid effector response, and participate in the generation of an adaptive immune response by modulating APC functions.84
DCs are professional APCs that confer early protection against pathogens and display a capacity to stimulate naive T cells and induce a specific immune response or tolerance, acting as a bridge between innate and adaptive immunity.85,86 In addition, they have recently been shown to be involved in supporting innate immunity by interacting with various innate lymphocytes, such as natural killer, natural killer T or TCR-gamma delta cells.87
DC precursors develop in the bone marrow and migrate through the blood into tissues, particularly at the mucosal surfaces and skin, where cells home and differentiate into resident populations that continuously sample the microenvironment for foreign substances and apoptotic host cells88. DCs can be divided into several subsets on the basis of cell-surface marker expression, maturity and function89. Although many subtypes arise from different developmental pathways, their phenotype and function are mainly modulated by signals that the cells receive from pathogens, the environment and from other immune cells.46,90–92
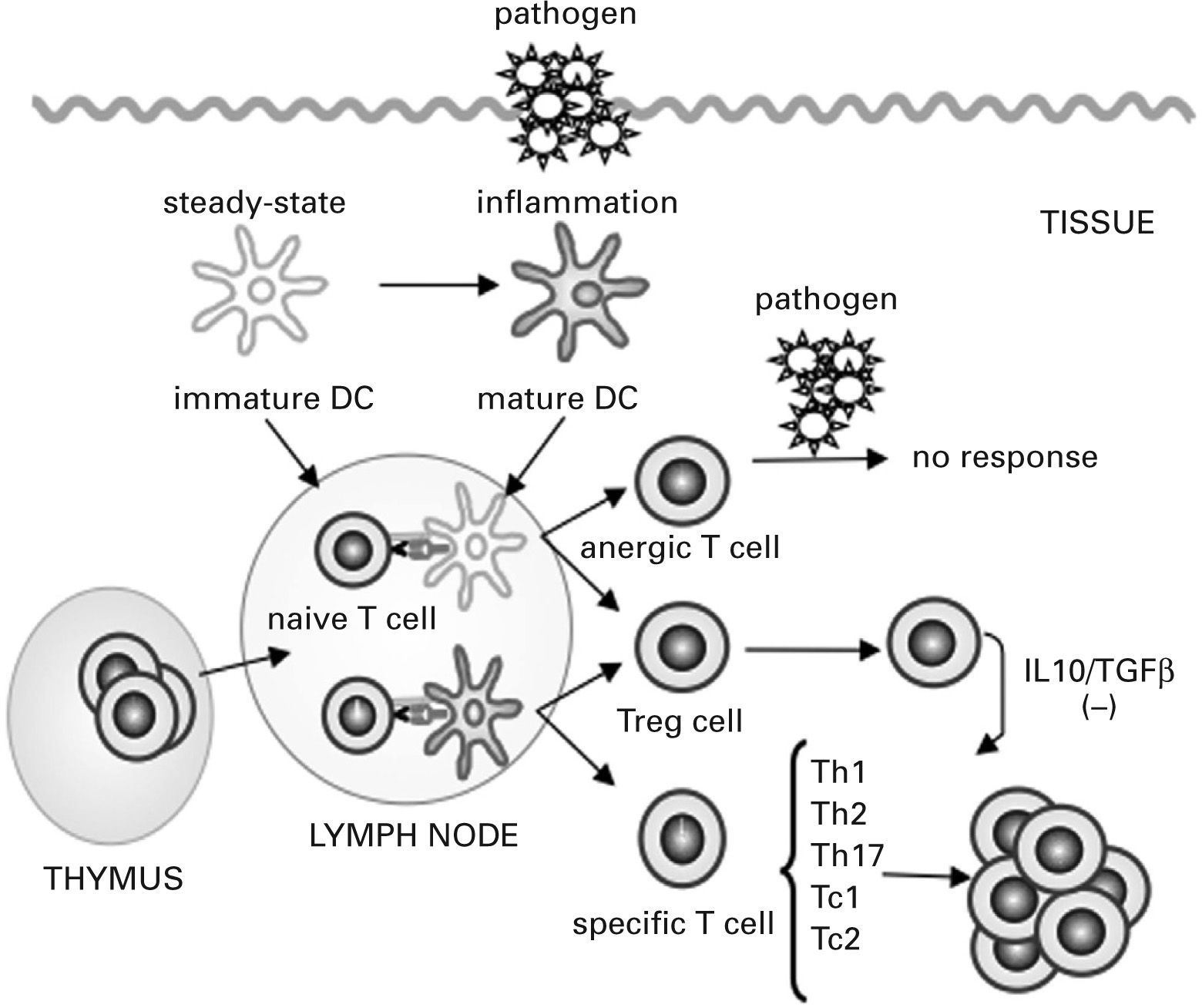
Under steady-state conditions, tissue-resident DCs are mostly immature, characterized by a high endocytic and phagocytic capacity, a low level of expression of MHC class II molecules and costimulatory molecules and a poor capacity to produce cytokines. Antigen presentation by these immature DCs leads to tolerance by inducing specific naive T-cell anergy or deletion and through the generation of T regulatory (Treg) cells, a group of T cells that actively suppress effector Th cells and cytotoxic T cells93–95 (see fig. 2). Alterations in Treg cell function have been associated with the pathogenesis of various disorders including autoimmunity, allergy, cancer, and infection with persistent pathogens96.
![Dendritic cells control tolerance and immunity in the periphery. In steady-state conditions, dendritic cells (DCs) remain immature but may still capture the antigen and migrate to the lymph nodes to prime naive cells and induce their apoptosis or anergy. Under specific conditions immature/ semi-mature DCs induce naive T cells to become specific T regulatory (Treg) cells. In the presence of signals generated by infection and injury, DCs change to a mature form and prime naive T cells for different effector functions (T helper [Th] 1, Th2, Th17, cytotoxic T [Tc] 1 and Tc2) and generate specific Treg cells that suppress T cell effector functions by releasing interleukin (IL) 10 and transforming growth factor β (TGFβ) or by direct contact.](https://static.elsevier.es/multimedia/03010546/0000003600000003/v1_201304101113/S0301054608725424/v1_201304101113/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNe5umqYt0zKZP4SYniTZ7+SgZrkCgayp3MRA1ovxa2vUxRfI/WuR9U+nfb86qvfSC2x9bCgRRKcLvZaNrbo0E9g9yLSGJZDzj7tZydev1N3kXatJWI2w23oWHZaMAl8Mx+B+JyMIEcsa6Hydj6X0seUMSZhTblAFjxBCK/PJ49q6A04wgTNG8HqTIHv+k2/6E/AU5Lzly+qEaU4Dc7MRFUpXjSo+MKHMBLkv58OacLPeLDizlpfqsdhLx5NHy2sBfTo/JI8k7GPnjFdzJSzFR1h "Dendritic cells control tolerance and immunity in the periphery. In steady-state conditions, dendritic cells (DCs) remain immature but may still capture the antigen and migrate to the lymph nodes to prime naive cells and induce their apoptosis or anergy. Under specific conditions immature/ semi-mature DCs induce naive T cells to become specific T regulatory (Treg) cells. In the presence of signals generated by infection and injury, DCs change to a mature form and prime naive T cells for different effector functions (T helper [Th] 1, Th2, Th17, cytotoxic T [Tc] 1 and Tc2) and generate specific Treg cells that suppress T cell effector functions by releasing interleukin (IL) 10 and transforming growth factor β (TGFβ) or by direct contact.")
Dendritic cells control tolerance and immunity in the periphery. In steady-state conditions, dendritic cells (DCs) remain immature but may still capture the antigen and migrate to the lymph nodes to prime naive cells and induce their apoptosis or anergy. Under specific conditions immature/ semi-mature DCs induce naive T cells to become specific T regulatory (Treg) cells. In the presence of signals generated by infection and injury, DCs change to a mature form and prime naive T cells for different effector functions (T helper [Th] 1, Th2, Th17, cytotoxic T [Tc] 1 and Tc2) and generate specific Treg cells that suppress T cell effector functions by releasing interleukin (IL) 10 and transforming growth factor β (TGFβ) or by direct contact.
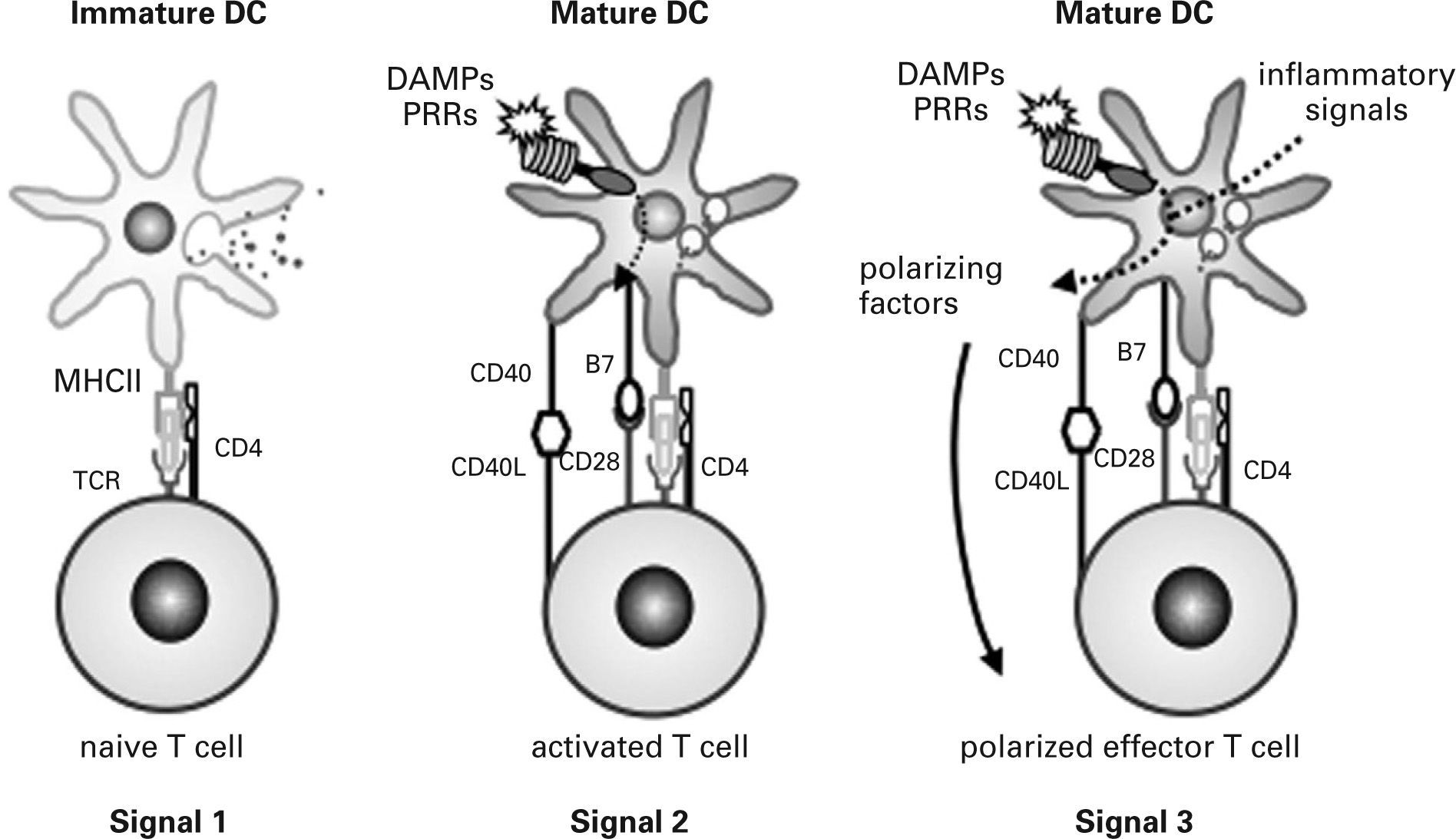
In infectious processes, immature DCs migrate to the injured region where they detect pathogens and damage via PRRs and receive other environmental inflammatory signals. Initially, immature DCs respond to injury by releasing inflammatory mediators that activate effector cells of the innate immune system. They then change their morphology, lose their phagocytic role and activate the antigen-processing machinery. During the maturation process, DCs express high levels of MHC and costimulatory molecules, reprogram their expression of chemokine receptors and acquire their T cell-polarizing capacity86,90,91,97. Simultaneously, DCs move to local lymph nodes and migrate into the T cell area, where they finally encounter naive antigen-specific T cells. DCs provide naive T cells with the three signals required for their activation and functional differentiation98 (fig. 3). Signal one is supplied through the TCR after recognition of antigen-specific peptides presented by DCs through MHC molecules. This recognition stimulates naive T cells and guarantees the specificity of the response but is insufficient to induce T cell proliferation and differentiation into effector cells. Signal two, the so-called costimulatory signal, is mediated by binding of different CD28 ligands (CD80 and CD86) expressed on DCs to CD28 on the naive T cell. This second signal stabilises the immune synapse through those adhesion molecules and CD40/CD40L activation. Signal three is mediated during antigen presentation by the binding of polarizing cytokines and other molecules released by mature DCs to their respective receptors on naive T cells. It has recently been shown that DCs also use the Notch pathway to polarize T cell differentiation99,100.
 to naive T cells determine their function: signal 1 guarantees specificity of response, signal 2 contributes to costimulation and stabilisation of the immune synapse and signal 3 leads to functional polarization of T cells. DAMPs, damageassociated molecular patterns; PRRs, pattern recognition receptors; TCR, T cell receptor.")
Activation of naive T cells. Three signals provided by dendritic cells (DC) to naive T cells determine their function: signal 1 guarantees specificity of response, signal 2 contributes to costimulation and stabilisation of the immune synapse and signal 3 leads to functional polarization of T cells. DAMPs, damageassociated molecular patterns; PRRs, pattern recognition receptors; TCR, T cell receptor.
According to the density and nature of the antigenic peptide presented, the class of co-stimulatory molecules expressed and the type of polarizing signals received, the naive CD4 + T cell differentiates to display a Th1, Th2, Th17 or Treg cell phenotype. DCs that secrete high levels of IL-12 p70 and IL-27 and express the notch ligand Delta-4, and those that produce type I interferons, instruct CD4 + naive cells to differentiate into Th1 cells101,102, whereas DCs that do not secrete high levels of IL-12 and express the notch ligand Jagged have the capacity to induce a Th2 response103,104. In addition the expression of OX40 ligand on DCs has been implicated in triggering the development of Th2 cells103. The differentiation of Treg cells is mediated by immature DCs but also by DCs that secret IL-10 and TGF-β and express the ICOS ligand103,105. In addition, TGF-β and IL-6 appear to act as polarising cytokines for the recently identified Th17 cells whereas IL-23 allows the activation of already differentiated Th17106. Environmental signals released by neighbouring cells are able to influence various function of DCs. For example, a novel cytokine, thymic stromal lymphopoietin, produced by human epithelial, stromal and mast cells exerts an effect on DCs that promotes specific Th2 cell differentiation and seems to be involved in the pathophysiology of inflammatory arthritis as well as allergic disease107,108. Other molecules that promote a Th2 response are prostaglandin E2,109, vitamin D110 and glucocorticoid111. Induction of Treg cells is modulated by molecules such as vasoactive intestinal polypeptide112 and vitamin A113. In addition DCs undergo an instructional phase induced by IFNγ and IL-10 released from surrounding cells114,115.
Maturation of DCs and the production of polarising signals is tightly regulated by pathogen recognition through PRRs in association with inflammatory signals and with other mediators released by innate lymphocytes and by local somatic cells46,97,98,116. Signals mediated through TLRs stimulate transcription of cytokines and chemokines, induce up-regulation of surface costimulatory molecules, and also affect antigen capture, processing and presentation, DC migration, and cell survival92,117. TLR activation is involved in the production of the polarizing cytokines IL-12118,119 and IL-10120,121, in the induction of the notch ligand Delta-like-4 on DC122 and in the release of thymic stromal lymphopoietin64. Recent evidence has suggested that signals from other PRRs could contribute to the elicitation of antigen-specific immunity116. Stimulation of the intracellular bacterial sensor NOD2 programs DCs to promote IL-17 production in human memory T cells123. It has been found that Nod1 stimulation can independently induce antigen-specific immunity with a predominant Th2 polarization profile, while in cooperation with TLR stimulation it leads to the development of Th1, Th2, and Th17-mediated immunity124.
Taken together, these findings suggest a high degree of functional plasticity of DCs to polarize naive T cells according to the type of pathogen and the presence of a variety of surrounding tissue factors.
FUTURE PERSPECTIVESThe recent evidence that PRRs play an essential role in innate and adaptive immune responses and the remarkable progress made in our understanding of their signalling pathways open a wide array of possibilities for the development of innovative therapies to treat infections and a long list of inflammatory diseases116,125. Suppression or modulation of TLR signalling by introducing natural ligands, soluble receptors, suppressors or antagonist proteins, and the use of genetic techniques that correct PRR signalling defects represent tools that could be used for the development of new immunotherapeutic strategies. The high level of functional plasticity displayed by DCs according to the signals generated through PRRs and environmental factors renders them attractive therapeutic targets for pharmacological redirection of the adaptive immune response, opening up new hopes for the design of cures for infections, cancer, allergy, and autoimmune diseases and in the induction of transplantation tolerance.54,126–131
In addition, therapies based on reducing IL-1 activity, such as the use of a recombinant form of the human IL-1 receptor antagonist (anakinra), have just begun and bring new hopes for the treatment of inflammatory syndromes associated with inflammasome dysfunction132–134. Other possible therapies such as anti-IL-1β monoclonal antibodies, the IL-1 Trap, IL-1 receptor type I antibodies, antibodies to the IL-1 receptor accessory chain and inhibitors of Caspase-1 will also be useful therapies in the future135.
It will be possible to use these new immunotherapies as individualized treatments in each patient according to the pathophysiology of the disease. Therefore, the appropriate prescription of these “biological agents” will necessitate a precise immunogenetic diagnosis and the development of techniques to allow reliable identification of the underlying cause of the disease as part of day-to-day clinical practice. Given the high degree of complexity of the processes that regulate the innate immune response and the cross-talk between the different signalling pathways, as well as the role of innate immunity in the development of acquired immunity, drastic changes in one of these immune mechanisms could give rise to undesirable side effects, as has been observed with some existing biological therapies136–139. Therefore, while these new therapies are promising, appropriate prescription and monitoring will require the establishment of multidisciplinary teams including immunology specialists.
I am grateful to Dr. I. Patten for editorial advice.
